Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
247 Cards in this Set
- Front
- Back
|
Platelet transfusion in the following circumstances is contraindicated EXCEPT:
A. Thrombotic Thrombocytopenia Purpura B. Renal Failure Thromobocytopathy C. Heparin Induced Thrombocytopenia D. Immune Thrombocytopenia Purpura E. Hemolytic-Uremic Syndrome |
Answer: B. Renal failure thrombocytopathy may be helped by platelet transfusion. TTP, HUS, and HIT are
absolute contraindications, but ITP is a relative contraindication because it just won’t help. |
|
|
Platelet transfusion in the following circumstances is contraindicated EXCEPT:
A. Thrombotic Thrombocytopenia Purpura B. Renal Failure Thromobocytopathy C. Heparin Induced Thrombocytopenia D. Immune Thrombocytopenia Purpura E. Hemolytic-Uremic Syndrome |
Answer: B. Renal failure thrombocytopathy may be helped by platelet transfusion. TTP, HUS, and HIT are
absolute contraindications, but ITP is a relative contraindication because it just won’t help. |
|
|
CHO antigens, naturally occuringnAB, AB ususally IgM, agglutination AB, react at immediate spin
|
ABO, Le, M,N,P
|
|
|
Protein Ag, acquired only after exposure, usually IgG, reactive at 37 degrees, coating AB
|
Rh,Kidd, Kell, S,s, and Duffy
|
|
|
Which one of the following disease-HLA associations is incorrect?
A. Type I diabetes mellitus – DR3, DR4 B. Ankylosing spondylitis – B27 C. Rheumatoid arthritis – DR4 D. Goodpasture’s disease – B35 E. Sjogren syndrome – DR3 |
Goodpasture’s disease is associated with DRB1*1501.
|
|
|
Which one of the following is not a characteristic component of DiGeorge’s syndrome?
A. hypocalcemia B. deletions on chromosome 11q22 C. cleft palate D. thymic aplasia E. cardiac abnormalities |
B. The characteristic genetic defects identified in DiGeorge’s syndrome are deletions at 22q11. The acronym
CATCH-22 (cardiac defects, abnormal facies, thymic hypoplasia, cleft palate, hypocalcemia, and 22q11 deletions) is helpful in recalling the components of the severe end of the clinical spectrum of this syndrome. |
|
|
Which one of the listed factors from the complement system is deficient in hereditary angioedema?
A. C1 B. C1 inhibitor C. C3 convertase D. properdin E. C4 |
Deficiency of C1 inhibitor (C1INH) is associated with hereditary angioedema, an inherited immune system
abnormality caused by low levels or dysfunctional C1 inhibitor. |
|
|
Ischemia-modified albumin reflects myocardial ischemia, rising within minutes of ischemic damage and returning to baseline within a few hours. The assay is based on the altered binding of albumin to which of the following elements?
calcium cobalt phosphorus oxygen iron |
COBALT.
The amino terminus of albumin is modified with exposure to a number of conditions, such as acidosis, hypoxemia, and free radicals. The modification decreased the ability of albumin to bind cobalt. The amount of unbound cobalt reflects the level of ischemia-modified albumin. |
|
|
Which of the following natriuretic peptides provides the most longitudinal information about congestive heart failure?
atrial (A-type) natriuretic peptide (ANP) brain (B-type) natriuretic peptide (BNP) pro-BNP N-terminal pro-BNP C-type natriuretic peptide |
N-TERM-PRO-BNP
A-type natriuretic peptide is released in response to increased ventricular as well as increased atrial filling pressures. For that reason, it's not as specific as BNP, which is only released in response to increased ventricular filling pressure (stretch). It is released as an inactive pro-BNP peptide which when cleaved releases an active BNP as well as the regulatory N-term-pro-BNP, a very stable molecule. Little is known about C-type natriuretic peptide. |
|
|
All of the following are included in the definition of acute coronary syndrome (ACS), except:
stable angina unstable angina congestive heart failure acute myocardial infarction sudden cardiac death |
CONGESTIVE HEART FAILURE.
All are considered to be in a spectrum of disease. Stable angina is reproducible and most likely due to progressive stenosis of coronary arteries. Unstable angina may represent further stenosis, but with a little less predictable event as well, such as a transient clot or vasospasm, causing transient ischemia. Acute MI is the best characterized and is the condition that we can most readily identify. |
|
|
What is the purpose of serial measurements of elevated troponins in suspected acute myocardial infarction?
A. increased sensitivity B. increase negative predictive value C. increased specificity A & B A, B, C |
INCREASED SENSITIVITY.
The specificity of a single elevated troponin is very high; serial measurements don't change that. However, a mildly elevated troponin many not be sensitive enough to detect AMI. Serial measurements of troponin increase the sensitivity, but don't change the ability of the test to change its negative predictive value. |
|
|
What is the purpose of measuring CK-MB in the presence of elevated troponin in a patient with a suspected acute myocardial infarction (AMI)?
troponin is less sensitive than CK-MB for AMI troponin is less specific than CK-MB for AMI troponin is not helpful in determining the time course of AMI CK-MB is more stable than troponin and stays elevated longer CK-MB can provide additional information about congestive heart failure |
TROPONIN IS NOT HELPFUL IN DETERMINING THE TIME COURSE OF AMI.
Troponin rises more slowly and stays elevated longer than CK-MB. If CK-MB continues to rise, it may indicate an acute event or an extension of an existing infarction, while a downward trend of CK-MB may indicate resolution of an infarction. |
|
|
In the quantitation of protein by the Kjedahl technique, what is actually measured?
spectrophotometry colorimetric assay refractometry ammonium nitrogen released by acid digestion |
AMMONIUM NITROGEN RELEASED BY ACID DIGESTION.
All of the techniques presented are used to measure protein. The Kjedahl technique is cumbersome and makes assumptions about average nitrogen content. Colorimetric assays are preferred for the measurement of protein, and all involve formation of a colored precipitate under alkaline or acidic conditions and then measuring the absorbance at the appropriate wavelength. Refractometry is used but has many interferences. Dye-binding is limited by uneven dye uptake by proteins. |
|
|
What is the usual net charge on proteins and toward which pole do they migrate?
negative, anode negative, cathode positive, anode positive, cathode no charge, it depends |
NEGATIVE, ANODE.
Most proteins bear a net negative charge at physiologic pH and as such migrate toward the anode or positive pole when subject to an electromotive force. Remember, anions have negative charges and are attracted to the positive pole or anode. Cations bear positive charges and are attracted to the negative pole or cathode. |
|
|
Which represents the fastest migrating band on standard serum protein electrophoresis performed at pH 8.6?
albumin alpha-1 alpha-2 beta gamma |
ALBUMIN.
Albumin accounts for the majority of normal serum protein and is the fastest migrating major protein followed by the alpha, beta, then gamma region proteins. |
|
|
All of the following techniques are used to characterize a suspected monoclonal band, except:
immunofixation electrophoresis immunotyping immunoelectophoresis immunoprecipitation all of the above are routinely used |
IMMUNOPRECIPITATION.
While immunoprecipitation can be used to characterize proteins, it is not commonly used to characterize monoclonal proteins. All the other techniques, especially immunofixation, are commonly used to identify and classify monoclonal proteins identified by serum electrophoresis. |
|
|
Which of the following condition(s) account for the most significant changes in serum albumin levels?
A. protein-losing enteropathy B. nephrotic syndrome C. iver disease D. A & B E. A, B, C |
A & B.
Protein-losing conditions are responsible for the greatest decrements in serum albumin. The ability of the liver to synthesize albumin is preserved with decreases only being apparent with severe end-stage liver disease. |
|
|
All of the following are functions of pre-albumin, except:
binding thyroid hormones, T3 & T4 bind and carry retinol-binding protein:vitamin A complex amyloid precursor in senile cardiac amyloidosis maintenance of serum osmotic pressure all of the above are functions of pre-albumin |
MAINTENANCE OF SERUM OSMOTIC PRESSURE.
Pre-albumin does function in the capacity to bind thyroid hormone, vitamin A, and is the precursor in cardiac amyloidosis and familial amyloid polyneuropathy. Albumin, rather than pre-albumin, is responsible for maintenance of serum osmotic pressure. Pre-albumin is prominent in the CSF. |
|
|
Which of the following descriptions best fits myoglobin?
most sensitive, least specific marker of acute myocardial infarction most specific, least sensitive earliest marker of acute myocardial infarction A & C B & C |
A & C.
Usually within moments of an acute MI, there is an elevation of the myoglobin. Unfortunately, very sensitive myoglobin is very nonspecific and can be elevated due to a number of causes. |
|
|
Ischemia-modified albumin reflects myocardial ischemia, rising within minutes of ischemic damage and returning to baseline within a few hours. The assay is based on the altered binding of albumin to which of the following elements?
A. calcium B. cobalt C. phosphorus D. oxygen E. iron |
COBALT.
The amino terminus of albumin is modified with exposure to a number of conditions, such as acidosis, hypoxemia, and free radicals. The modification decreased the ability of albumin to bind cobalt. The amount of unbound cobalt reflects the level of ischemia-modified albumin. |
|
|
Which of the following natriuretic peptides provides the most longitudinal information about congestive heart failure?
A. atrial (A-type) natriuretic peptide (ANP) B. brain (B-type) natriuretic peptide (BNP) C. pro-BNP D. N-terminal pro-BNP E. C-type natriuretic peptide |
N-TERM-PRO-BNP
A-type natriuretic peptide is released in response to increased ventricular as well as increased atrial filling pressures. For that reason, it's not as specific as BNP, which is only released in response to increased ventricular filling pressure (stretch). It is released as an inactive pro-BNP peptide which when cleaved releases an active BNP as well as the regulatory N-term-pro-BNP, a very stable molecule. Little is known about C-type natriuretic peptide. |
|
|
All of the following are included in the definition of acute coronary syndrome (ACS), except:
stable angina unstable angina congestive heart failure acute myocardial infarction sudden cardiac death |
CONGESTIVE HEART FAILURE.
All are considered to be in a spectrum of disease. Stable angina is reproducible and most likely due to progressive stenosis of coronary arteries. Unstable angina may represent further stenosis, but with a little less predictable event as well, such as a transient clot or vasospasm, causing transient ischemia. Acute MI is the best characterized and is the condition that we can most readily identify. |
|
|
What is the purpose of serial measurements of elevated troponins in suspected acute myocardial infarction?
increased sensitivity increase negative predictive value increased specificity A & B A, B, C |
INCREASED SENSITIVITY.
The specificity of a single elevated troponin is very high; serial measurements don't change that. However, a mildly elevated troponin many not be sensitive enough to detect AMI. Serial measurements of troponin increase the sensitivity, but don't change the ability of the test to change its negative predictive value. |
|
|
What is the purpose of measuring CK-MB in the presence of elevated troponin in a patient with a suspected acute myocardial infarction (AMI)?
troponin is less sensitive than CK-MB for AMI troponin is less specific than CK-MB for AMI troponin is not helpful in determining the time course of AMI CK-MB is more stable than troponin and stays elevated longer CK-MB can provide additional information about congestive heart failure |
TROPONIN IS NOT HELPFUL IN DETERMINING THE TIME COURSE OF AMI.
Troponin rises more slowly and stays elevated longer than CK-MB. If CK-MB continues to rise, it may indicate an acute event or an extension of an existing infarction, while a downward trend of CK-MB may indicate resolution of an infarction. |
|
|
In the quantitation of protein by the Kjedahl technique, what is actually measured?
spectrophotometry colorimetric assay refractometry ammonium nitrogen released by acid digestion |
AMMONIUM NITROGEN RELEASED BY ACID DIGESTION.
All of the techniques presented are used to measure protein. The Kjedahl technique is cumbersome and makes assumptions about average nitrogen content. Colorimetric assays are preferred for the measurement of protein, and all involve formation of a colored precipitate under alkaline or acidic conditions and then measuring the absorbance at the appropriate wavelength. Refractometry is used but has many interferences. Dye-binding is limited by uneven dye uptake by proteins. |
|
|
What is the usual net charge on proteins and toward which pole do they migrate?
negative, anode negative, cathode positive, anode positive, cathode no charge, it depends |
NEGATIVE, ANODE.
Most proteins bear a net negative charge at physiologic pH and as such migrate toward the anode or positive pole when subject to an electromotive force. Remember, anions have negative charges and are attracted to the positive pole or anode. Cations bear positive charges and are attracted to the negative pole or cathode. |
|
|
Which represents the fastest migrating band on standard serum protein electrophoresis performed at pH 8.6?
albumin alpha-1 alpha-2 beta gamma |
ALBUMIN.
Albumin accounts for the majority of normal serum protein and is the fastest migrating major protein followed by the alpha, beta, then gamma region proteins. |
|
|
All of the following techniques are used to characterize a suspected monoclonal band, except:
immunofixation electrophoresis immunotyping immunoelectophoresis immunoprecipitation all of the above are routinely used |
IMMUNOPRECIPITATION.
While immunoprecipitation can be used to characterize proteins, it is not commonly used to characterize monoclonal proteins. All the other techniques, especially immunofixation, are commonly used to identify and classify monoclonal proteins identified by serum electrophoresis. |
|
|
Which of the following condition(s) account for the most significant changes in serum albumin levels?
protein-losing enteropathy nephrotic syndrome liver disease A & B A, B, C |
A & B.
Protein-losing conditions are responsible for the greatest decrements in serum albumin. The ability of the liver to synthesize albumin is preserved with decreases only being apparent with severe end-stage liver disease. |
|
|
All of the following are functions of pre-albumin, except:
binding thyroid hormones, T3 & T4 bind and carry retinol-binding protein:vitamin A complex amyloid precursor in senile cardiac amyloidosis maintenance of serum osmotic pressure all of the above are functions of pre-albumin |
MAINTENANCE OF SERUM OSMOTIC PRESSURE.
Pre-albumin does function in the capacity to bind thyroid hormone, vitamin A, and is the precursor in cardiac amyloidosis and familial amyloid polyneuropathy. Albumin, rather than pre-albumin, is responsible for maintenance of serum osmotic pressure. Pre-albumin is prominent in the CSF. |
|
|
Transferrin may be elevated with iron deficiency and resemble an M-spike on serum protein electrophoresis. Where does the transferrin band migrate?
pre-albumin albumin alpha-1 alpha-2 beta-1 |
BETA-1.
Transferrin is the predominant beta-1 protein. On standard serum electrophoresis, beta does not resolve into beta-1 and beta-2, but can on high-resolution electrophoresis. (In the beta-2 region migrates IgA, C-reactive protein can be in beta-2 or gamma-2). The predominant alpha-1 band is alpha-1-antitrypsin; alpha-2 has haptoglobin and ceruloplasmin. |
|
|
This protein is elevated in serum with renal or hepatic disease:
ceruloplasmin alpha-2-macroglobulin haptoglobin transferrin fibrinogen |
ALPHA-2-MACROGLOBULIN.
Due to its large size, alpha-2-macroglobulin is typically not lost with nephrotic syndrome. As a result of the loss of other smaller proteins and fluid, the alpha-2-macroglobulin concentration increases. |
|
|
The asialated form of this protein is also known as tau protein and can be found in cerebrospinal fluid:
pre-albumin albumin transferrin alpha-1-antitrypsin ceruloplasmin |
TRANSFERRIN.
Both the unmodified and asialated forms of transferrin can cross the blood-brain barrier through active transport. This accounts for the double transferrin peak typically seen in CSF electrophoresis. |
|
|
This protein should not normally be found in serum, but when it is, it runs with the beta-globins:
C-reactive protein fibrinogen haptoglobin ceruloplasmin alpha-2-macroglobulin |
FIBRINOGEN.
Normally blood clots to make serum, and fibrinogen is consumed. In the event of incomplete clotting (such as in heparinized patients), fibrinogen may appear at the beta-gamma interface. |
|
|
What is the clinical significance of a twin albumin band?
M-spike normal variant acute inflammation starvation high cholesterol |
NORMAL VARIANT.
Bisalbuminemia is a normal variant seen in heterozygotes for different albumin allotypes. There is no clinical significance. |
|
|
In non-selective proteinuria, all of the bands on serum protein electrophoresis are decreased, except:
albumin alpha-1 alpha-2 beta gamma |
ALPHA-2.
Albumin is usually the most commonly affected protein. Due to its small size, it is lost in both selective and non-selective proteinuria. Other proteins also start to decrease in the serum in non-selective proteinuria, except alpha-2-macroglobulin, due to its large size. |
|
|
Beta-gamma bridging is most commonly seen in which of the following situations?
monoclonal gammopathy cirrhosis starvation non-selective proteinuria selective proteinuria |
CIRRHOSIS.
Predominantly due to increased IgA, beta-gamma bridging is seen with cirrhosis. Cirrhosis can also show hypoalbuminemia with blunted alpha-1 and alpha-2 peaks. |
|
|
All of the following are potential causes of apparent hypogammaglobulinemia, except:
congenital hypogammaglobulinemia lymphoma nephrotic syndrome myeloma all of the above are potential causes of hypogammaglobulinemia |
ALL OF THE ABOVE ARE POTENTIAL CAUSES OF HYPOGAMMAGLOBULINEMIA.
It may be counter-intuitive, but a significant portion of cases of myeloma can exhibit hypogammaglobulinemia, especially when there is a concomitant Bence-Jones protein seen on urine protein electrophoresis. |
|
|
All of the following are characteristic features of normal CSF protein electrophoresis relative to serum protein electrophoresis, except:
oligoclonal gamma bands prominent pre-albumin band dim albumin band double beta-transferrin band dim alpha-2 band |
OLIGOCLONAL GAMMA BANDS.
Oligoclonal bands, though they can be present in CSF protein electrophoresis, are distinctly NOT normal. Oligoclonal bands in CSF but not in a concurrent SPEP support a diagnosis of multiple sclerosis. |
|
|
Which of the following types of proteinuria presents with a strong albumin band on urine protein electrophoresis?
tubular proteinuria glomerular proteinuria tubulointerstitial proteinuria overflow proteinuria none of the above patterns exhibit a strong albumin band |
GLOMERULAR PROTEINURIA.
Glomerular proteinuria occurs due to a loss of the selective filtration of the glomerulus - large proteins, such as alpha-2 macroglobulin are retained while very small proteins are resorbed in the tubules, leaving medium-sized proteins, such as albumin in the urine. Tubular proteinuria is due to the loss of small protein resorption, while overflow proteinuria is due to very high serum levels of a protein overwhelming the kidney's filtration and resorption capacity. |
|
|
Which of the following types of cryoglobulins is most commonly associated with Waldenstrom macroglobulinemia?
Type I Type II Type III Type IV Type V |
TYPE I.
Type I cryoglobulins are monoclonal, associated with monoclonal gammopathies, such as myeloma or Waldenstrom macroglobulinemia. Type II is a mixture of polyclonal IgG and monoclonal IgM, while Type III is a mixture of two or more polyclonal antibodies. |
|
|
What's the most common cause of mixed cryoglobulinemia?
chronic liver disease lupus hepatitis C virus lymphoproliferative disorders chronic infections |
HEPATITIS C VIRUS.
Prior to the advent of HCV testing, approximately 1/2 of all cases of mixed cryoglobulinemia had no apparent cause. With testing, the majority of these cases were found to be due to HCV. |
|
|
What's the most common renal pathology associated with mixed cryoglobulinemia?
A. membranoproliferative glomerulonephritis, type II B. membranoproliferative glomerulonephritis, type I C. membranous glomerulonephritis D. focal segmental glomerulosclerosis E. minimal change disease |
MEMBRANOPROLIFERATIVE GLOMERULONEPHRITIS, TYPE II.
Cryoglobulinemia is a deposition disease leaving dense immune complex deposits within the mesangium and subendothelium, often with the characteristic tubuloreticular inclusions or “interferon fingerprints.” There is often an associated vasculitis. |
|
|
What is the most common adverse effect of correcting hyponatremia too slowly?
central pontine myelinolysis cerebral edema cardiac arrhythmias anesthesia reflex hypoglycemia |
CEREBRAL EDEMA.
Carefully read the question if you chose central pontine myelinolysis. That most often occurs when hyponatremia is corrected too rapidly. Hyponatremia is usually described as a serum sodium level of less than 135mM. |
|
|
All of the following are potential causes of hypervolemic hyponatremia, except:
cardiac failure nephrotic syndrome cirrhosis renal tubular acidosis, type I all of the above lead to hypervolemic hyponatremia |
RENAL TUBULAR ACIDOSIS, TYPE I.
The causes of true hyponatremia (relatively low sodium compared to water) can be subdivided according to the patient's volume status (assessed clinically). Hypovolemic patients tend to be hyponatremic due to water loss either through the kidneys or GI tract. Euvolemic patients are often hyponatremic due to drugs and similar conditions. Hypervolemic patients are hyponatremic due to the “oses” - cardiosis, nephrosis, and cirrhosis, better known as congestive heart failure, nephrotic syndrome, and cirrhosis. |
|
|
Which of the following are potential causes of hypokalemia?
GI potassium losses renal potassium losses transcellular shifts all of the above none of the above |
ALL OF THE ABOVE.
Measurement of urinary potassium helps to distinguish renal potassium losses from other causes. Transcellular shifts come into play most often with correction of diabetic ketoacidosis. As the hyperglycemia of ketoacidosis is corrected, the concomitant hyperkalemia is often overcorrected as potassium moves back into cells. For that reason, it is important to include supplemental potassium when correcting the hyperglycemia of diabetic ketoacidosis. |
|
|
Which of the following causes of acidosis is associated with hypokalemia?
A. renal tubular acidosis, type I B. renal tubular acidosis, type II C. renal tubular acidosis, type IV D. A & B E. A, B, C |
A & B.
Type I, or distal renal tubular acidosis, is due to the inability to produce an acid urine. Type II, or proximal renal tubular acidosis, is due to bicarbonate wasting. Both are associated with hypokalemia. There is no Type III RTA. Type IV renal tubular acidosis is due to aldosterone deficiency and is associated with hyperkalemia. |
|
|
What is the cause of primary hyperparthyroidism?
excess parathyroid hormone CASr gene mutation thiazide diuretics sarcoidosis hyperthyroidism |
EXCESS PARATHYROID HORMONE.
Straightforward question - too much PTH. The most common cause is a parathyroid adenoma, followed by parathyroid hyperplasia and carcinoma, |
|
|
Which of the following causes of hypercalcemia is least likely to be associated with kidney stone formation?
A. sarcoidosis B. hypervitaminosis D C. primary hyperparathyroidism D. milk-alkali syndrome E. squamous cell carcinoma-associated PTHrP |
PRIMARY HYPERPARATHYROIDISM.
Most causes of hypercalcemia also cause hyperphosphatemia. The combination of high calcium and high phosphate leads to increased calcium phosphate crystal formation and deposition. Crystals are often deposited in the kidney, leading to stones or in vessels with severe end-stage renal disease (calciphylaxis). |
|
|
What accounts for the majority of cases of hypercalcemia?
A. hyperparathyroidism B. malignancy C. hypervitaminosis D D. A & B E. A, B, C |
A & B.
80-90% of hypercalcemia is due to hyperparathyroidism or malignancy (often with the paraneoplastic expression of PTHrp). PTH is synthesized as an intact protein then digested into three fragments. Intact and N-terminal PTH have biological activity and, as a result, short half-lives. |
|
|
Which of the following equations best estimates the anion gap?
A. [HCO3 −] + [K+] − [Na+] B. ([Na+] − [K+]) − [HCO3 −] C. ([Cl−] − [HCO3 −]) + [Na+] D. [HCO3 −] − ([Na+] − [Cl−]) E. [Na+] − ([Cl−] + [HCO3 −]) |
E. [NA + ] − ([CL − ] + [HCO 3 − ]).
|
|
|
Which of the following crystals is most strongly birefringent?
calcium pyrophosphate monosodium urate hydroxyapatite corticosteroid cholesterol |
MONOSODIUM URATE.
Monosodium urate is the most strongly birefringent crystal seen in synovial fluids. Urate crystals are needle-shaped with negative birefringence and rapid extinction (goes away quickly with a small change in the angle of the compensator) under polarization. Negatively birefringent crystals are yellow when parallel to the long axis of the compensator and blue when perpendicular. Positive birefringence is the opposite. Urate crystals are needle-shaped and a needle looks like a minus sign (negative birefringence). Also, yellow and parallel have double “l's.” See the text for an additional helpful mnemonic. |
|
|
Which of the following criteria is considered positive for a diagnostic peritoneal lavage?
>1 mL gross blood RBCs >1000/mL WBCs >5/mL DPL fluid in Foley catheter mesothelial cells >100/mL |
DPL FLUID IN FOLEY CATHETER.
More than 15mL of blood, 100,000 red blood cells/mL, 500 white blood cells/mL are considered positive findings. Finding lavage fluid in the places other than the peritoneum, such as in the bladder or pleural space, is also considered a positive finding. Finally, if any bacteria is present on gram stain, that is considered a positive finding. |
|
|
Which of the following tests are characteristically elevated in a pleural effusion associated with rheumatoid arthritis?
pH glucose LDH A & B A, B, C |
LDH.
Typically, the pH and glucose are decreased, while the LDH and rheumatoid factor levels are elevated. |
|
|
Which feature most specifically distinguishes a parapneumonic pleural effusion from an empyema?
the presence of neutrophils specific gravity >1.010 LDH levels the absence of bacteria acidic pH |
THE ABSENCE OF BACTERIA.
An empyema is usually the result of a secondarily-infected parapneumonic effusion. Neutrophils can be present in a parapneumonic effusion, but not to the levels seen in an empyema. The pH of an empyema tends to be acidic. The most important differentiation factor is the presence or absence of bacteria. |
|
|
Which of the following pleural fluid test results confirms your clinical suspicion of an exudate?
pleural fluid to serum protein ratio >0.3 pleural fluid to serum LDH ratio >0.3 pleural fluid LDH >200, or 2/3 the upper limit of serum LDH specific gravity >1.006 pleural fluid protein >1 g/L |
PLEURAL FLUID LDH >200, OR 2/3 THE UPPER LIMIT OF SERUM LDH.
The Light criteria are the most commonly used guidelines for the determination of whether an effusion is an exudate or transudate. Once the fluid is categorized, then potential etiologies can be explored. A protein ratio >0.5, an LDH ratio >0.6, and LDH >200 are all suggestive of an exudate. Some other ancillary tests can also be performed to help with determination, such as specific gravity (>1.01 implies exudate, also protein >3g/dL, cholesterol >45 mg/dL, and a bilirubin ratio >0.6). |
|
|
Confirmed positive test for HbsAg or repeatedly reactive test for anti-HBc
Laboratory evidence of HCV infection Laboratory evidence of HTLV-1 infection Have donated the only unit of blood to a patient who developed HIV or HTLV and had no other probable cause of infection |
Permanent deferrals
|
|
|
Travelers to variant Creutzfeld-Jacob areas
Use of bovine insulin manufactured in UK History of babesiosis of Chagas disease Etretinate (Tegison) Receiving money or drugs for sex(prostitute) |
Permanent deferrals
|
|
|
History of syphilis or gonorrhea, treatment for syphilis or gonorrhea, or positive syphilis screening test
Receipt of blood products, human tissue, or plasma-derived clotting factors Hepatitis B immune globulin administration |
One year deferrals
|
|
|
Application of tattoo
Mucous membrane exposure to blood Nonsterile skin penetration Residing with or having sexual contact with an individual with viral hepatitis |
One year deferrals
|
|
|
Being incarcerated for >72 consecutive hours
Travelers to malaria-endemic areas(after departure, if symptom free, regardless of prophylaxis) Paying for sex |
One year deferrals
|
|
|
Dutasteride (Avodart)
|
6 months after last dose
deferral for donation |
|
|
Besides OSHA, which of the following federal agencies regulates laboratory operations?
A.Federal Bureau of Investigation B. Central Intelligence Agency C. Department of Transportation D. Social Security Administration |
C. The Department of Transportation regulates laboratory operations related to safe practices in packaging, transporting, and handling biologic materials.
Require training q 2 yr for those involved in shipping samples |
|
|
JCAHO
|
Accredits hospitals; inspects every 3 yrs (starting in 2006 on an unannounced basis)
|
|
|
COLA
|
Accredits laboratories only; inspects every 2 yr with voluntary self-assessment
Mostly inspects MD office labs and smaller labs, some hospital labs |
|
|
CLIA established requirements for retention of records and AP materials
|
For most, (such as blocks, instrument printouts, CP reports, QC records, etc., )requirement is 2 years
For cytology slides, 5 years minimum For AP slides and reports, 10 years minimum |
|
|
BASIC, COBOL, MUMPS - Programming languages for writing programs; MUMPS developed for medical software
|
BASIC, COBOL, MUMPS - Programming languages for writing programs; MUMPS developed for medical software
|
|
|
Neutralization of Antibodies for Lewis, H
|
Saliva (secretor for Leb)
|
|
|
Neutralization of Antibodies for P1
|
Hydatid cyst fluid,
Pigeon egg fluid |
|
|
Neutralization of Antibodies for Sda
|
Urine,guinea pig
|
|
|
Neutralization of Antibodies for chido,
Rodgers |
Serum
|
|
|
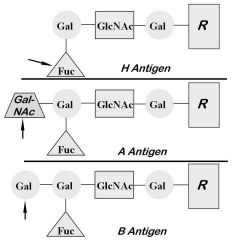
Se gene: Enzyme adds fucose to type I chains at terminal
galactose; product is H antigen. |
|
|
H gene: Enzyme adds fucose to terminal galactose of
type II chains, in blood; product is H antigen |
|
|
Genotypes: AA, AO
Antigens: A, H Antibodies: anti-B (primarily IgM) Dolichos biflorus agglutinates A1 |
Group A
|
|
|
Genotypes: BB, BO
Antigens: B, H Antibodies: Anti-A (primarily IgM). |
Group B
|
|
|
Least frequent ABO blood type (about 4%)
Antigens: A and B (very little H) Antibodies: none |
Group AB
|
|
|
Total lack of H, A, and B antigens
Develop strong anti-H, anti-A, and anti-B O forward, O reverse, but antibody screen positive |
Bombay (Oh) Phenotype
|
|
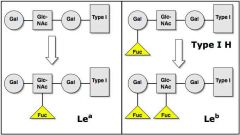
|
In secretors, Se product adds fucose, then Le
product adds fucose; this makes Leb (Lewis B) 1) So, the majority of secretor’s chains are Leb |
|
|
P Blood Group (the cool one)
|
also built on ABO-related chains
P1 most frequent antigen |
|
|
P Blood Group (the cool one)
|
rare people lack all three antigens and make anti-PP1Pk and develop acute HTR, early abortions
|
|
|
Wiener’s Haplotypes
R1: DCe r’: dCe R2: DcE r”: dcE R0: Dce r: dce Rz: DCE ry: dCE |
“R” = D, “r” = d
• “1” or “prime” = C • “2” or “double prime” = E • “0” or “blank” = ce • Any superscript letter = CE |
|
|
“The Big Four”
Whites: R1 > r > R2 > R0 Blacks: R0 > r > R1 > R2 |
“The Big Four”
Whites: R1 > r > R2 > R0 Blacks: R0 > r > R1 > R2 |
|
|
Citrate-phosphate-dextrose (CPD) and citratephosphate-
dextrose-dextrose (CP2D) |
Allows 21 days of RBC/Whole Blood storage
Used before additive solutions |
|
|
Citrate-phosphate-dextrose-adenine (CPDA-1)
|
Very similar to CPD but with 17.3 mg of adenine (no adenine in CPD)
Allows 35 days of RBC/Whole Blood storage |
|
|
Additive solutions
Increases shelf life of RBCs to 42 days |
Most common types
1) AS-1 (Adsol®) + mannitol 2) AS-3 (Nutricel®) 3) AS-5 (Optisol®) +mannitol c. Specifics vary, but all add more dextrose and adenine to increase blood shelf life |
|
|
Allows blood to be stored for extended periods
without drastic effects on most metabolic and therapeutic qualities |
|

|
Blood is a controlled product that is tightly regulated
by the Food and Drug Administration (with many regulations from the American Association of Blood Banks and College of American Pathologists) |
|
|
Volume: 450-500 ml
Contents: RBCs (200 ml) Plasma (250 ml) WBCs (109) and platelets Anticoagulant (63 or 70 ml) |
Whole Blood
|
|
|
Red Blood Cells with and without additives
|
Volume: ~250 ml (350 ml with additive solutions)
Contents: RBCs (~200 ml); HCT < 80% Plasma (50 ml with CPDA-1) WBCs (108) and platelets Anticoagulant Additive solution (if applicable) 200 mg iron |
|
|
Platelet Concentrate (PC, “random platelets”)
|
Volume: 50 ml
Contents: Platelets (>5.5 x 1010 in 90% tested) Plasma (including ~80 mg fibrinogen) WBCs (107) pH at or over 6.2 |
|
|
Thrombocytopathy
|
Metabolic effects (renal or hepatic failure)
• Remember, though, that platelets are not first-line defense against platelet-related bleeding in renal failure! (Think DDAVP, Cryo, conjugated estrogens, etc) |
|
|
Volume: ~ 100 ml
Contents: Platelets (>3.0 x 1011 in 90% tested) Plasma (incl ~150 mg fibrinogen) WBCs (106-108) |
Apheresis platelets (“single donor” platelets)
|
|
|
Leukocyte reduction
|
1) Leukocyte Reduced Red Cells
a) At least 85% of original RBCs and < 5 x 106 white cells in 95% of tested units |
|
|
Leukocyte reduction
|
2) Leukocyte Reduced Platelet Concentrate
a) At least 5.5 x 1010 platelets in 90% of units tested, and < 8.3 x 105 WBCs in 95% of tested units |
|
|
Leukocyte reduction
|
3) Leukocyte Reduced Apheresis Platelets
a) At least 3.0 x 1011 platelets in 90%, and < 5 x 106 WBCs in 95% of tested units |
|
|
Fresh Frozen Plasma (FFP)
|
Volume: 200-250 ml
Contents: All coagulation factors • 400 mg fibrinogen • 1 IU/ml of all others Almost no viable cells Anticoagulant |
|
|
Volume: Approximately 15 mL
Contents: > 150 mg fibrinogen (us. ~250 mg) > 80 IU factor VIII 80-120 IU von Willebrand’s Factor 40-60 IU factor XIII Fibronectin |
Cryoprecipitate
|
|
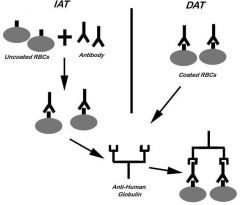
|
1. IAT: described above; checks for in-vitro coating of
RBCs with antibody or complement. 2. DAT: no 37 C incubation step; checks for in-vivo coating of RBCs with antibody or complement |
|
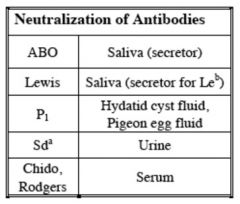
|
A particular substance, when mixed with an antibody,
eliminates the activity of that antibody against test red cells |
|
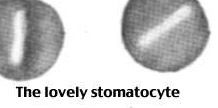
|
Rhnull phenotype
1) No Rh antigens whatsoever 2) Hemolytic anemia with stomatocytes |
|
|
Kidd Blood Group
|
Kidd antigens
a. Jka, Jkb b. Jka more common (exception to “Rule of B’s”) |
|
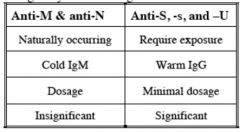
|
MNSs Blood Group
Glycophorin A carries M and N antigens Glycophorin B carries S, s, and U antigens |
|
|
MNSs Blood Group
|
MNSs antigens
a. M frequency roughly equals N b. s>S c. If S-s-, may also be U negative if black |
|
|
anti N induced by hemodialysis
|
formaldehyde sterilization of machine with modification of N antigen
|
|
|
High frequency: k (99.8%), Jsb, Kpb
Low frequency: K (9% whites, 2% blacks), Jsa, Kpa Kx: important antigen that helps stabilize RBC membrane Kell system antigens destroyed by thiol reagents (ZZAP, DTT) but untouched by enzymes alone. |
Kell antigens
|
|
|
Kell antibodies
|
Anti-K: most common non-ABO
antibody after anti-D Warm reacting IgG anti-k(analogous to anti-K) |
|
|
Kell Blood Group
|
Consequences of incompatibility
a. Severe HTRs 1) May be acute or delayed; usually extravascular. b. Severe HDN |
|
|
(Platelet countpost – Platelet countpre) x BSA//
Number of platelets transfused |
Interpretation
a. 7000-10,000 or above means adequate response b. Two consecutive inadequate CCI’s define refractoriness |
|
|
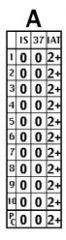
Warm autoantibodies (panel A)
a. Across-the-board positivity (at IAT +/- 37 C) with positive autocontrol (Panel “A” in chart above) b. Positive DAT c. Antibody specificity: Very broad, likely basic Rh component. |
|
|
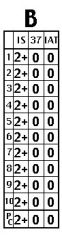
Cold autoantibodies (panel B)
a. Across-the-board positivity (at IS +/- 37 C) with positive autocontrol (panel “B” above) b. Positive DAT (usually for complement components only) c. Antibody specificity: usually I, sometimes i d. Strategy 1) Consider prewarmed crossmatches. 2) Consider transfusion through a blood warmer. |
|
|
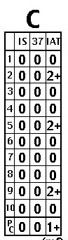
Antibodies vs. recently transfused antigens (panel
C) a. One or more antibodies in a panel with positive autocontrol and history of recent transfusion (panel “C” above) 1) Classic autocontrol description: “mixed field” a) Definition: two groups of RBCs, with/without agglutination. 2) Of most clinical importance when antibody screen was negative before transfusion b. Positive DAT (also “mixed field”) c. Famous with Kidd and Duffy antibodies d. Strategy 1) Ensure that the patient is stable clinically (rule out delayed hemolysis); support as necessary. 2) Phenotype transfused unit, if possible. 3) Give antigen negative blood in future. |
|

|
High-titer, low-avidity antibodies (HTLA) (panel
D) a. Classically 1+ positivity at AHG only, with negative autocontrol (panel “D” above) 1) Occasional HTLA’s can give positive autocontrol and DAT’s; uncommon b. Still positive after many dilutions (“high titer”) but weakly reacting (“low avidity”) c. Chido, Rodgers most common antigens 1) Complement components 2) Neutralize with serum d. Clinically insignificant (No HDN, no HTRs) e. NOTE: The pattern of this panel could also be seen in other high-frequency antibodies that may be significant! |
|
|
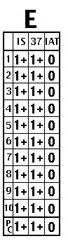
Reagent-related antibodies (panel E)
a. Antibodies against reagents used in testing (e.g., preservatives in LISS) b. Across-the-board positivity at IS/37 C, negative at IAT, positive autocontrol (panel “E” above) c. DAT negative (due to washing step) d. Run reactions without offending reagent. |
|
|
Recovered from malaria
Immigrants from malaria-endemic areas Acitretin (Soriatane) after last dose |
Three year deferrals
|
|
|
Live attenuated viral and bacterial vaccines: german measles and
chicken pox (varicella) Finasteride (Proscar, Propecia) after last dose Isotretinoin (Accutane) after last dose |
Deferrals Four Weeks:
|
|
|
Immunization Deferrals
Two Weeks: |
Measles
Mumps Oral Polio Yellow Fever Oral typhoid |
|
|
Immunization Deferrals
No Deferral: |
Hepatitis A and B
Influenza DPT Anthrax Pneumococcus Lyme Disease Cholera Polio (injection) Typhoid (injection) |
|
|
Physical Requirements; donor
|
Weight: > 110 lbs (50 Kg)
Temperature: < 99.5o F (37.5 C) Pulse: 50-100 bpm (unless athlete) Blood Pressure: < 180/100 Hemoglobin or Hematocrit: > 12.5 g/dl or 38% |
|
|
Infectious disease screening (as of March 2004)
|
1) HbsAg
2) Anti-HBc 3) Anti-HCV 4) HCV RNA by PCR (HCV Nucleic Acid Testing, or “HCV NAT”) a) HCV window period decreased from ~70 days with anti-HCV alone to between 10 and 30 days with HCV NAT 5) Anti-HIV-1,2 6) HIV RNA by PCR (HIV NAT) a) HIV window period decreased from 16 days with combination of anti-HIV and p24 to about 10 days with HIV NAT 7) Anti-HTLV-I, II 8) Serologic test for syphilis 9) HIV-1-Ag (p24); may delete if doing HIV NAT |
|
|
In which of the following reactions, is the pathology mediated by donor anti-HLA anti-neutrophil
antibodies attacking the recipient WBCs? A. Transfusion associated graft versus host disease B. Anaphylactoid reaction C. Transfusion related acute lung injury D. Febrile nonhemolytic transfusion reaction E. None of the above |
Answer: C. Transfusion associated acute lung injury (TRALI) is due to the donor anti-HLA antibodies
attacking the recipient WBCs. |
|
|
All of the following are associated with contamination of RBC units EXCEPT:
A. Yersinia enterocolitica B. E. coli C. Citrobacter freundii D. Pseudomonas species E. Gram positive cocci |
Answer: E. Gram positive cocci are associated with platelet transfusion.
|
|
|
IgA deficient patients are at risk for which of the following type transfusion reactions?
A. Febrile non-hemolytic transfusion reaction B. Anaphylactic reaction C. Anaphylactoid reaction D. Febrile hemolytic transfusion reaction E. Urticarial hypersensitivity reaction |
Answer: B. Anaphylactic reactions are classically described in IgA deficient patients. Anaphylactoid
reactions are usually described in the context of someone on an ACE inhibitor (bradykinin effect). |
|
|
The missense mutation Ser810Lys in the mineralocorticoid receptor results in:
a. Neoplastic processes unresponsive to hormonal therapy b. Dominant form of pseudohypoaldosteronism c. Early-onset hypertension that is exacerbated by pregnancy d. Early-onset hypertension that is not accelerated by pregnancy e. A constitutively activated receptor with abnormal activation by aldosterone |
c. Patients with the
Ser810Lys mutation have severe hypertension , which present mostly before age 20. The mutation is not associated with neoplastic formation. The dominant forms of pseudohypoaldosteronism are secondary to the following inactivating mutations: ΔG1226, ΔT1597, C/T1831stop, and ΔA intron splice site. The mutation results in a constitutively activated mineralocorticoid receptor; however, it retains its normal activation by aldosterone. |
|
|
An increased level of S-warfarin, under standard anticoagulant drug dosage, would be expected in a patient with which of the following polymorphisms?
a. CYP2C9*2 b. CYP2C9*3 c. CYP2C19*2 d. All of the above e. Only a and b are correct. |
e. Individuals
with CYP2C9*2 and CYP2C9*3 have an impaired metabolism of -warfarin , which leads to an increased plasma concentration of the drug despite standard dosing. Hence, such patients will experience an increased risk for serious or life-threatening bleeding complications despite obtaining a standard dose of medication. The VKORC1 polymorphism likewise has a role in warfarin therapy. |
|
|
Which of the following does NOT support the Hardy-Weinberg law of population genetics?
a. It enables one to have the ability to take information about the frequency of alleles in the population and make predictions about the frequency of genotype in the population. b. Individuals with all genotypes are equally capable of mating and passing on their genes (there is no selection against any particular genotype). c. Rate of mutation will not have an appreciable effect. d. The population tested is large and matings are expected to be random with respect to the locus in question. e. There is no significant immigration of individuals from a population with allele frequencies markedly different from the endogenous population. |
c. Genetic equilibrium is a basic principle of population genetics. Violations of the
Hardy-Weinberg law can cause deviations from expectation. Among such deviations are inbreeding/co-sanguinuity (increases homozygosity for all genes), small population size (can cause random change in genotypic frequency - also known as genetic drift), and assortative mating (causes an increase of homozygosity only of those genes involved in the trait). The law further assumes that there is no appreciable rate of mutation; changes in this manner would affect the allelic frequencies, which are assumed to be constant. |
|
|
Which of the following NAT1 gene variants retains normal enzyme activity?
a. NAT1*1 b. NAT1*2 c. NAT1*8 d. NAT1*14 e. NAT1*15 |
a. All polymorphisms with a “star” *1 are considered to be wild-type by convention. Hence, all other variants are altered, leading to either excess, depressed, or absent production.
|
|

What type of structural chromosomal abnormality is identified in ?
a. Insertion b. Duplication c. Derivative chromosome d. Isochromosome e. Paracentric inversion |
e. Paracentric inversion
consists of an inversion within a given arm of the chromosome. Insertions “insert” additional chromosome, whereas duplication inserts a portion of duplicated chromosome. Isochromosomes lose one of the chromosome arms and contains a duplicate inverted other chromosome arm (e.g., 2p arms in mirror image at centromere). |
|
|
most severe vWD (homozygous for the defective gene) and may have severe mucosal bleeding, no detectable vWF antigen, and may have sufficiently low factor VIII that they have occasional hemarthroses (joint bleeding), as in cases of mild hemophilia.
|
Type 3
|
|
|
autosomal dominant type of vWD caused by gain of function mutations of the vWF receptor on platelets; specifically, the alpha chain of the glycoprotein Ib receptor (GPIb). This protein is part of the larger complex (GPIb/V/IX) which forms the full vWF receptor on platelets.
The ristocetin activity and loss of large vWF multimers is similar to type 2B, but genetic testing of VWF will reveal no mutations. |
Platelet-type(also known as pseudo-vWD or platelet-type (pseudo) vWD)
|
|
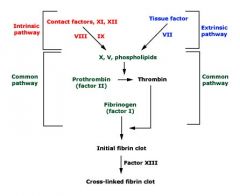
|
Intrinsic (and common) pathway is assessed (aPTT) and the extrinsic (and common) pathway by the (PT). The thrombin time (TT) assesses the final step in the common pathway, the conversion of fibrinogen to fibrin, following the addition of exogenous thrombin. Fibrin is crosslinked through the action of factor XIII, making the final fibrin clot insoluble in 5 Molar urea or monochloroacetic acid
|
|
|
12,11,9,8, 10, 5, 2,1 (PTT)
7(PT) |
12,11,9,8, 10, 5, 2,1 (PTT)
7(PT) |
|
|
GP IIb/IIIa
|
fibrinogen & VWF receptor
|
|
|
GP Ib/V/IX
|
receptor for VWF
|
|
|
ADP, epi, collagen, TXA2
testing? |
GPIIb/IIIa
fibrinogen |
|
|
Ristocetin
testing? |
GPIb
VWF |
|
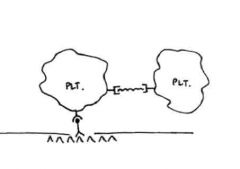
The image for this question shown on the website represents initial platelet adhesion to the
vessel wall and aggregation. What structure is represented by the red box in the image? A. GPIb receptor B. GPIIb/IIIa receptor C. Fibrinogen D. Collagen E. Von Willebrands factor |
The image for this question shown on the website represents initial platelet adhesion to the
vessel wall and aggregation. What structure is represented by the red box in the image? A. GPIb receptor Answer: A. GPIb receptor on the platelet is responsible for initial adhesion of the platelet to the vessel wall via vWF as a bridge, which binds to exposed collagen. |
|
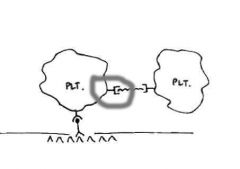
The image for this question shown on the website represents initial platelet adhesion to the
vessel wall and aggregation. What structure is represented by the red box in the image? A. GPIb receptor B. GPIIb/IIIa receptor C. Fibrinogen D. Collagen E. Von Willebrands factor |
The image for this question shown on the website represents initial platelet adhesion to the
vessel wall and aggregation. What structure is represented by the red box in the image? B. GPIIb/IIIa receptor Answer: B. GPIIb/IIIa receptor mediates aggregation of platelets with fibrinogen serving as the bridge between the GPIIb/IIIa receptors. |
|
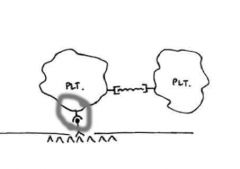
The image for this question shown on the website represents initial platelet adhesion to the
vessel wall and aggregation. What structure is represented by the red box in the image? A. GPIb receptor B. GPIIb/IIIa receptor C. Fibrinogen D. Collagen E. Von Willebrands factor |
The image for this question shown on the website represents initial platelet adhesion to the
vessel wall and aggregation. What structure is represented by the red box in the image? A. GPIb receptor |
|
|
Adhesion of platelets is mediated through all of the following EXCEPT:
A. GPIIb/IIIa B. GPIb C. Collagen D. von Willebrand's factor |
Answer: A. GPIIb/IIIa receptor on platelets is responsible for aggregation. The GPIIb/IIIa receptor binds
with fibrinogen to create a platelet plug. GPIb, collagen, and von Willebrand's factor all work in concert together to cause adhesion of the platelet to the vessel wall. When the endothelium of the vessel is disrupted, collagen is exposed which binds to von Willebrand's factor. Von Willebrand's factor serves as a bridge between the collagen and the GPIb receptor on the platelet. |
|
|
The PFA-100 analyzer has replaced the bleeding time at many institutions. This test utilizes two
tubes for analysis. One tube contains collagen and ADP, while the other tube has collagen epinephrine. In a patient who has a prolonged bleeding time, which of the following would have a normal clotting time in the collagen and ADP tube but be prolonged and the collagen epinephrine tube? A. Von Willebrand's disease B. Thrombocytopenia C. Bernard-Soulier disease D. Aspirin effect E. All the above will affect both tubes |
Answer: D. Aspirin results in a thromboxane deficiency. In the PFA-100 analyzer, a thromboxane
deficiency is overcome by the abundance of ADP. Therefore this test can be helpful in separating storage pool defects, which include aspirin, from other possible at realities. That being said, bleeding times and the PFA-100 are still not very sensitive or specific tests. |
|
|
Which of the following would be least helpful in treating a patient with hemophilia A:
A. DDAVP B. Recombinant factor VIII C. FFP D. Cryoprecipitate E. All of the above would be helpful |
C. FFP contains very little factor VIII needed to increase the patient’s levels. Cryoprecipitate is
rich in vWF and Factor VIII (remember they travel together). DDAVP releases factor VIII and vWF from endothelial cells. |
|
|
A patient underwent cardiac bypass surgery and was having an uneventful post-operative course. On
POD #7, the patient developed an ischemic leg. The intern asks your help in diagnosis, and also informs you that his platelet count has recently dropped. The most likely etiology is: A. DVT with paradoxical embolus B. Heparin induced thrombocytopenia C. Thrombotic thrombocytopenia D. Idiopathic Thrombocytopenia purpura E. Hyperhomocystinemia |
B. Heparin induced thrombocytopenia (HIT) usually develops approximately 5-10 after starting
treatment with heparin (time period can be shorter for patients with previous exposure). In cases of HIT with significant risk of thrombosis the platelet count usually drops >50%. HIT results from antibodies against heparin bound to PF4 on platelets. The platelet count usually recovers in 2-5 days after discontinuing heparin. (Goodnight, pages 425-431) |
|
|
Which of the following tests would be most helpful in diagnosing the patient described in question 5?
A. Quantitative d-dimer B. Qualitative d-dimer C. Heparin antibody D. ADAM-TS level E. Methyltetrahydrofolate reductase mutation |
C. Heparin antibody
|
|
|
A patient who is known to have von Willebrand’s disease (vWD) has a normal vWF antigen and
decreased levels of Factor VIII. What is the most likely vWD subtype in this patient? A. Platelet type, pseudo-von Willebrand’s disease B. vWD, type 2A C. vWD, type 2B D. vWD, type 2M E. vWD, type 2N |
E. vWD type 2N (Normandy) is characterized by normal amounts of vWF, which interact with
platelets in a normal fashion. They have a mutation in the region that binds with Factor VIII, which results in decreased affinity. Factor VIII is degraded in the plasma approximately five time faster than when protected by vWF. Platelet type, pseudo-von Willebrand’s disease looks identical to vWD type 2B, except the mutation is a gain of function in the GPIb receptor where as vWD type 2B is a gain of function in the vWF. |
|
|
A patient is suspected of having Bernard-Soulier disease. If the patient has this disease, they would
show response to all of the following during platelet testing EXCEPT: A. ADP B. Epinephrine C. Collagen D. Ristocetin E. Thromboxane A2 |
D. Bernard-Soulier disease is defined by deficiency of the GpIb receptor on the platelet. GpIb is
responsible for platelet adhesion, which is stimulated only by ristocetin in platelet testing. |
|
The diagram shown for this question illustrates initial platelet adhesion and aggregation. The
substance highlighted by the red box best represents? A. Von Willebrand's factor B. GPIIb/IIIa C. GPIb D. Fibrinogen E. Fibrin |
D. The highlight red box represents fibrinogen which is responsible for the initial aggregation of
platelets mediated via the GPIIb/IIIa receptors. Von Willebrand's factor links to expose collagen to platelets via the GPIb receptor. |
|
|
All of the following may result in a prolonged thrombin clotting time (TCT) EXCEPT:
A. Factor II deficiency B. Heparin contamination C. Dysfibrinogenemia D. Hypofibrinogenemia E. Increased fibrin degradation products |
A. The thrombin clotting time (TCT) measures the conversion of fibrinogen to fibrin. The test
supplies thrombin (factor II) to start this process. Heparin contamination, abnormal fibrinogen (i.e. dysfibrinogenemia), low fibrinogen levels (usually < 100), fibrin degradation products, and high concentrations of immunoglobulin (especially IgM – stays intravascular) can cause prolongation of the TCT. |
|
|
Of extracellular matrix constituents, which is the most important pro-thrombotic component?
A. Collagen B. Proteoglycans C. Fibronectin D. Adhesive gylcoproteins E. None of the above |
A. Collagen is the most important pro-thrombotic component of the extra cellular matrix. Von
Willebrand’s factor acts as a bridge between collagen and platelets. |
|
|
A 46 y/o woman is found to have a prolonged aPTT with a normal PT. A mixing study was
performed, and the aPTT corrected into the high normal range. Clinically, the patient does not have any evidence of bleeding or bleeding tendencies. Further questioning reveals the patient has been noted to have a prolonged aPTT during a routine physical exam 5 years ago. He was referred to a hematologist at that time who told him he was fine and not to worry. Based on these findings, which of the following is the most likely etiology of the patients prolonged aPTT? A. Factor VIII deficiency (mild hemophilia A) B. Factor IX deficiency (hemophilia B) C. Factor XII deficiency D. Lupus anticoagulant E. Factor V inhibitor |
C. In vitro factor XII is required to bind to glass beads to activate the intrinsic clotting pathway.
This factor is not required to activate the clotting cascade in vivo, and patients with deficiencies of factor XII, high molecular weight kininogen, or prekallikrein do not have a bleeding diathesis clinically. A mild hemophilia A might be a reasonable answer, but the patient is a woman, and hemophilia A follows an Xlinked inheritance pattern. Lupus anticoagulant and factor V inhibitors would not result in a corrected mixing study. |
|
|
An acquired inhibitor to factor X can be caused by which of the following?
A. Use of bovine thrombin B. Amyloidosis C. Severe Hemophilia A D. Severe Hemophilia B E. None of the above |
B. Amyloidosis is a cause of an acquired inhibitor to factor X. Hemophilia A patients may
develop Factor VIII inhibitors, and Hemophilia B patients may develop inhibitors to factor IX. Use of bovine thrombin in vascular surgery is associated with development of factor V inhibitors. |
|
|
Which of the following has the greatest effect in inhibiting both factors V and VIII?
A. Anti-thrombin III B. Thrombomodulin C. Factor XIII D. Thromboplastin E. Tissue factor pathway inhibitor |
B. Thrombomodulin acts on thrombin to activate Protein C (with Protein S as a cofactor) to cause
proteolysis of factors V and VIII. Anti-thrombin III inhibits activity of thrombin and factors XIIa, XIa, Xa, and IXa. It is potentiated by heparin. Factor XIII is platelet stabilizing factor. Thromboplastin is another name for tissue factor, and tissue factor pathway inhibitor complexes with factor Xa and VIIa. |
|
|
Heparin induced thrombocytopenia is caused by heparin interacting with:
A. Anticardiolipin antibodies B. Platelet factor 4 C. GpIIb/IIIa D. HLA receptors E. None of the above |
B. HIT is caused by platelet factor 4 interactions with heparin.
|
|
|
All of the following are symptoms of type 1 von Willebrand’s disease EXCEPT:
A. Easy bruising B. Joint bleeding C. Oral bleeding D. Epistaxis |
B. Joint bleeding is characteristic of Hemophilia A (Factor VIII deficiency). Type 2N is
characterized by abnormal binding of vWF to Factor VIII, which results in a hemophilia A-like picture. |
|
|
Which of the following best represents the three steps of normal hemostasis?
decreased heart rate, adhesion of platelets, plug formation fibrin plug, inflammation, hypotension heat, redness, swelling vasoconstriction, platelet aggregation, fibrin formation vascular damage, stasis, endothelial injury |
D. VASOCONSTRICTION, PLATELET AGGREGATION, FIBRIN FORMATION.
Initially, vascular damage is met with the body's reaction of “clamping down” to avoid further blood loss. This usually takes the form of vasoconstriction. In response, platelets adhere to vessels, then aggregate with each other. The primitive platelet plug (alliteration!) is then replaced by the fibrin clot produced through the action of the clotting cascade. |
|
|
All of the following are components of platelet alpha granules, except:
von Willebrand Factor platelet-derived growth factor serotonin platelet factor-4 P-selectin |
C. SEROTONIN.
The P compounds (PDGF, P-selectin, PF4) and vWF, along with a number of other proteins, such as IGF-1 and TGF beta, are all found in the alpha granules of platelets. These granules give the platelets their distinctive purple color with Wright-Giemsa staining. Deficiencies of alpha granules lead to grey platelet syndrome. |
|
|
Which platelet surface antigen acts as the receptor for fibrinogen?
GPIb/V/IX ADP receptor GPIIb/IIIa GPIa/IIa GPIc/IIa |
C. GPIIB/IIIA.
The GPIb/V/IX complex (CD42) serves as the receptor for platelet adhesion through vWF. GPIIb/IIIa then comes to assist with platelet aggregation through the binding of fibrinogen. Deficiencies in Ib lead to Bernard-Soulier syndrome, while mutation of GPIIb/IIIa accounts for the weakened (“thrombasthenia”) platelet binding in Glanzmann. |
|
|
What is the primary use of the point of care clotting time assay?
monitoring dialysis effectiveness monitoring coumadin therapy monitoring heparin therapy screening test for von Willebrand disease screening test for activated protein C resistance |
C. MONITORING HEPARIN THERAPY.
The activated clotting time assay is used primarily to monitor heparin in patients on supratherapeutic amounts of heparin where the PTT may exceed the reportable range. Since it is a point-of-care test, it is rapid and most useful in pre-operative or pre-procedure (dialysis) settings. |
|
|
What is the usual effect on coagulation testing in the presence of elevated Factor VIII levels?
prolong PT shorten PT prolong PTT shorten PTT no change in either PT or PTT |
SHORTEN PTT.
Factor VIII, a constituent of the intrinsic pathway, is most sensitively monitored by PTT. The PTT is responsive to changes in factors involved in either the intrinsic or common pathways. For this reason, elevated Factor VIII causes a shortening of the PTT. |
|
|
All of the following can cause prolongation of both PTT and PT, except:
deficiencies of Factors X, V, and II disseminated intravascular coagulation liver disease anti-Factor VIII antibodies vitamin K deficiency |
D. ANTI-FACTOR VIII ANTIBODIES.
An antibody against Factor VIII presents as a prolongation of aPTT with a relatively normal PT due to Factor VIII being required for the intrinsic pathway but not the extrinsic pathway of coagulation. |
|
|
What ratio of patient plasma to normal plasma is recommended for the detection of weak inhibitors of coagulation?
1:1 2:1 4:1 1:2 1:4 |
C. 4:1.
Weak inhibitors can be overwhelmed with too much normal plasma. For that reason, it is recommended that a smaller amount is used. The greater ratio has shown to have a much greater sensitivity for detecting inhibitors while remaining relatively specific. |
|
|
A mutation in this coagulation factor accounts for activated protein C (APC) resistance:
thrombin Factor V Factor VIII Factor X Protein C |
B. FACTOR V.
Inhibition of coagulation by Protein C involves the cleavage of Factor V at a conserved sequence. The mutation of that site on Factor V (Leiden) results in a protein that is resistant to degradation and cleavage by activated Protein C. Hence, the patient is at risk for thrombosis. |
|
|
What's the most common cause of antiphospholipid syndrome?
lupus anticoagulant anti-phospholipase antibody anti-Factor Xa antibody anti-cardiolipin antibody Factor V Leiden |
D. ANTI-CARDIOLIPIN ANTIBODY.
The vast majority of cases of antiphospholipid syndrome are due to anticardiolipin antibody with the remainder of cases due to lupus anticoagulant. |
|
|
. What is the purpose of the anti-Factor Xa assay?
routine workup of elevated aPTT routine workup of elevated PT monitoring low molecular weight heparin alternative to PT for monitoring coumadin indirect assay of protein C levels |
C. MONITORING LOW MOLECULAR WEIGHT HEPARIN.
Low molecular weight and unfractionated heparin are difficult to monitor. Since they selectively inhibit Factor X, the aPTT is less reliable than it is for monitoring heparin, which also inhibits Factor II. Anti-Xa provides an alternative assay. |
|
|
mild to moderate reduction of vWF, most common and mildest
|
Type 1vWD
|
|
|
The absence of high molecular weight (HMW) VWF results in a loss of platelet-dependent function
|
Type 2AvWD
|
|
|
Defective VWF causes overactive binding of A1 platelet receptor, resulting in a lack of high molecular weight multimers
|
Type 2BvWD
|
|
|
Similar to type 2B, but platelets do not clump; not associated with multimer defects
|
Type 2MvWD
|
|
|
Defective VWF cannot bind to FVIII; similar results of mild hemophilia A, which can lead to misdiagnoses
|
Type2N
|
|
|
The rarest and most severe form of VWD; no detectable VWF levels, resulting in lower factor VIII levels
|
Type 3vWD
|
|
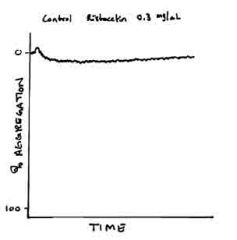
The diagram shown for this case represents a platelet control and the patient's platelets is
tested for aggregation in the presence of low dose and high-dose ristocetin. In addition, electrophoresis showed decreased large molecular weight von Willebrand's multimers. Mixing the patient's plasma with random donor platelets resulted in the same platelet arrogation findings with high and low dose ristocetin. Based on this information, the best diagnosis is: A. Bernard-Soulier syndrome B. Glanzman's thromboblasthenia C. Pseudo-von Willebrand's disease D. von Willebrand's disease, type 2B E. Cannot be determined with the given information |
D. This case illustrates an example of font Willebrand's disease, type 2B. vWD type 2B is
characterized by a gain of function mutations in the vWF which leads to spontaneous binding of platelets and rapid clearance of high molecular weight multimers. Pseudo-von Willebrand's disease is also in the differential diagnosis and is characterized by a gain of function of the GPIb receptor. Given that is a defect of the platelet, mixing the patient's plasma with random donor platelets would expect to correct the abnormal aggregation study with low and high-dose ristocetin, which does not happen in this case. |
|
|
How does GPIb become activated in vivo and in vitro, respectively?
shear force, ristocetin ristocetin, compression activation of ADP receptor, ristocetin binding vWF, epinephrine fibrin, fibrin |
A. SHEAR FORCE, RISTOCETIN.
GPIb along with GPV/IX acts as a receptor for vWF on the exposed basement membrane of the endothelium. Shear forces from the circulation activate the GPIb. Ristocetin is an antibiotic with the side effect of promoting platelet adhesion. |
|
|
Which protein crosslinks platelets through GPIIb/IIIa?
collagen fibrin Factor XIII Factor IIa antithrombin |
B. FIBRIN.
Thrombin released from platelet alpha granules activates fibrinogen by cleaving propeptide to yield fibrin, which, in addition to helping start coagulation, also contributes to platelet aggregation. |
|
|
Which factor is unique to the extrinsic pathway of coagulation?
II V VII IX X |
C. VII.
Factor VIIa functions as a “tenase,” activating Factor X, as well as a “ninase,” activating Factor IX, leading into the intrinsic pathway |
|
|
All of the following factors are inhibited by antithrombin, except:
thrombin Factor IXa Factor Xa Factor XIIa Factor Va |
FACTOR VA.
Antithrombin functions mainly by inactivating thrombin (really!) and Factor Xa, a process that is facilitated by heparin. In addition to Factors II and X, antithrombin also inactivates Factors IX, XII, and XI. Factors V and VIII, on the other hand, are not enzymes, but rather cofactors, inhibited by the action of Protein C and catalyzed by Protein S. |
|
|
All of the following are routinely used in assaying platelet function by aggregometry, except:
ATP collagen epinephrine ristocetin arachidonate |
. ATP.
While arachidonate is used less often that the others, ATP is not used. ADP is used as a stimulant of aggregation through its receptor. Ristocetin is an antibiotic that stimulates adhesion. |
|
|
What does the secondary wave of platelet aggregation seen with the biphasic low-dose ADP and epinephrine response represent?
increased binding to collagen platelet degranulation increased activation by collagen costimulation by coagulation formation of fibrin dimers |
B. PLATELET DEGRANULATION.
Degranulation of platelet dense granules which are full of small molecules, such as ATP, Ca++, and serotonin, leads to further stimulation of platelet aggregation. Initial stimulation (first wave) is due to the direct action of low-dose ADP or low-dose epinephrine. |
|
|
Which of the following disorders is associated with normal platelet aggregation with all agonists except ristocetin?
von Willebrand disease Bernard-Soulier syndrome Glanzmann thrombasthenia A & B A, B, C |
D. A & B.
Glanzmann thrombasthenia is characterized by the opposite pattern - response to only ristocetin - and is due to mutation of the GPIIb/IIIa receptor. Bernard-Soulier is due to a defect in GPIa, while von Willebrand disease is due to defects in von Willebrand Factor, which, due to their interactions, can present with overlapping clinical symptoms. |
|
|
Which disorder is associated with diminished clot retraction?
grey platelet syndrome Glanzmann thrombasthenia storage pool defect Bernard-Soulier syndrome von Willebrand disease |
B. GLANZMANN THROMBASTHENIA.
Clot retraction requires functional GPIIb/IIIa receptor in order to begin wound healing after the process of thrombus formation has begun. Since clot retraction requires GPIIB/IIIa, it will be aberrant in individuals with Glanzmann thrombasthenia. |
|
|
The ristocetin induced platelet aggregation (RIPA) is an in vitro assay for von Willebrand factor activity[1] used to diagnose von Willebrand disease. It has the benefit over the ristocetin cofactor activity in that it can diagnose type 2B vWD and Bernard-Soulier syndrome.
|
ristocetin causes von Willebrand factor to bind the platelet receptor glycoprotein Ib (GpIb), so when ristocetin is added to normal blood, it causes agglutination.
In von Willebrand disease, where von Willebrand factor is absent or defective, abnormal agglutination occurs: In type 1 vWD: no agglutination occurs In type 2A vWD: no agglutination occurs In type 2B vWD: hyperactive agglutination occurs In type 2N vWD: normal agglutination occurs In type 3 vWD: no agglutination occurs |
|
|
All of the following are derived from myeloid stem cells, except:
A. NK cells B. histiocytes C. monocytes D. eosinophils E. dendritic cells |
NK CELLS.
Lymphocytes, including B, T, and NK cells, are all derived from a common lymphoid stem cell. Cells of the reticuloendothelial system, such as macrophages, histiocytes, and dendritic cells, as well as the granulocytes, monocytes, megakaryocytes, and erythrocytes, are all derived from myeloid precursors. |
|
|
What type of immunoglobulin receptor do mast cells express?
Fc alpha Fc beta Fc gamma Fc delta Fc epsilon |
FC EPSILON.
Mast cells are capable of binding IgE through the Fc epsilon receptor. |
|
|
Which chromosome bears the genes for the heavy chains?
2 22 14 16 it depends on which heavy chain you are talking about |
14.
The genes for mu, gamma, alpha, delta, and epsilon are all on the same chromosome. This is important for the mechanism of class-type switching. |
|
|
What is the next step in B cell development for a cell that produces a self-reactive immunoglobulin?
the heavy chain undergoes class-switching the light chain redoes VDJ recombination only the variable portion of the light chain is removed the variable portion undergoes somatic hypermutation to change its specificity the cell undergoes apoptosis and dies |
THE CELL UNDERGOES APOPTOSIS AND DIES.
Part of the high level of variety that is generated through the development of B cells is a subset of cells that produce self-reacting immunoglobulins. Alas, a cell that produces a self-reacting immunoglobulin is scheduled to undergo apoptosis and die. |
|
|
Which of the following stages is the last in B cell development to express CD34 and TdT?
lymphoid stem cell pro-B-cell pre-B-cell B cell plasma cell |
PRO B-CELL.
In the development of a pro-B-cell to a pre-B-cell, the immature markers CD34 and TdT are lost. The equivalent in the T cell is the transition from prothymocyte to mature thymocyte. Note that there is an intermediate stage of T-cell development, the immature thymocyte that still expresses TdT, but not CD34. |
|
|
Which of the following is required for isotype switching?
antigen stimulation Th stimulation migration from bone marrow to spleen A & B A, B, C |
D. A & B.
Isotype switching, the process whereby the IgM immunoglobulin originally produced is able to become IgG, IgA, or another subclass, requires both the surface IgM to bind to its cognate epitope as well as stimulation by helper T cells. There is no mass migration of isotype-switched B cells from the marrow to the spleen. That's just crazy. |
|
|
Which of the following antibodies activates complement through the alternative pathway?
IgG IgA IgM IgD IgE |
IGA.
There are only three isotypes of immunoglobulin capable of activating complement: IgG, IgA, and IgM. Of these three, only IgA activates complement through the alternative, rather than the classical pathway of complement activation. Of note are that there are subclasses of several immunoglobulins and that IgG3 is unable to activate complement. |
|
|
What additional signal is required for T cells to bind to antigen-presenting cells through the T cell receptor?
CD44 CD16 MHC IgE complement C3 |
C. MHC.
While immunoglobulin both soluble and on the surface of the B cell is able to recognize antigenic isotopes on presenting cells, the T cell receptor requires the antigens to be present complexed to either Class I MHC, which is expressed on almost all nucleated cells in the case of CD8+ T cells, or Class II MHC, which is present on dedicated antigen presenting cells and is recognized by CD4+ T cells. |
|
|
T cell receptor is presented on the T cell surface bound in a noncovalent fashion to this marker:
CD2 CD3 CD5 CD4 CD8 |
B. CD3.
As a member of the signaling complex with the T-cell receptor, CD3 helps transmit the activating signal when the TCR binds a MHC-bound antigen. |
|
|
Which of the following markers acts as the NK cell receptor for the constant region of the IgG heavy chain?
CD16 CD56 CD57 CD68 CD1a |
CD16.
Antigen-dependent cellular cytotoxicity (ADCC) works through the CD16 binding of IgG constant regions for immunoglobulin-coated opsonized cells. This accounts for one of the primary means that NK cells facilitate the removal of virus-infected and tumor cells. |
|
|
Which of the following antigen-presenting cells express S100 and CD1a?
Langerhans cell interdigitating reticulum cell of the interfollicular portion of lymph nodes monocyte-macrophage A & B A, B, C |
D. A & B.
When CD1a positivity is discussed, often Langerhans cells are considered alone in the differential diagnosis. But it is important to note (especially in the case of some neoplasms) that CD1a is not a specific marker for Langerhans cells. In fact, almost all “professional” antigen-presenting cells including Kuppfer cells in the liver, Hoffbauer cells in the placenta, and the dendritic reticulum cell found in lymph node germinal centers can all be CD1a (+). |
|
|
What cytokine secreted by T lymphocytes stimulates eosinophilic development?
IL-1 TNF alpha IFN gamma IL-5 IL-6 |
IL-5.
TH2 cells secrete IL-4 to stimulate the production of IgE and IL-5 to stimulate eosinophils, especially in response to parasite infection. |
|
|
Which of the following complement proteins is an opsonin, promoting opsonization through binding cells?
C1a C3b C3a C5a C9 |
C3B.
Both C3b and Ig act as opsonins, coating cells and targeting their destruction through several means, including through antigen-dependent cellular cytotoxicity (ADCC) pathway (IgG) or through direct lysis by the membrane attack complex of complement proteins (C3b). |
|
|
At what point do the classical and alternative pathways of complement activation coalesce?
the association of C4 with C2 the conversion of C3 to C3a and C3b the association of C4b2b with C3b the conversion of C5 to C5a and C5b they are completely separate at all times |
THE CONVERSION OF C5 TO C5A AND C5B.
The alternate and classical pathways of complement activation use different means and factors to achieve the same goal - the creation of a C5 convertase, which is the driving force behind the formation of the C5-C9 membrane attack complex. Some memorable complement factoids: The classical pathway most often is activated by IgG (with the notable exception of IgG4). The alternate pathway is most often directly activated. The alternate pathway relies on low levels of C3 convertase activity to make a C3 convertase of its own, whereas the classical pathway makes a C3 convertase de novo. |
|
|
Which of the following byproducts of complement activation function as anaphylatoxins increasing vascular permeability?
C5a C3a C2a A & B A, B, C |
D. A & B.
Some of the products of the complement cascade perform functions outside of their role in creating the membrane attack complex. C3b is an opsonin like IgG and both C2a and C5a function as anaphylatoxins. |
|
|
What chromosome is home to the major histocompatibility complex?
3 6 11 13 17 |
6.
MHC Classes I, II, and III are all located on the short arm of chromosome 6. Class I encodes receptors found on almost all nucleated cells. Class II encodes receptors found on antigen-presenting cells. Class III encodes a hodge-podge of important and varied proteins, such as 21-hydroxylase, HFE, and TNF-alpha. |
|
|
What is the chance that two siblings are of identical HLA haplotypes?
0% 25% 50% 75% 100% |
25%.
The MHC genes are extremely polymorphic, composed of numerous alleles. It is important to remember that despite the size of the complex, there is very rare crossing over and recombination and therefore the entire region is inherited as a haplotype (barring sporadic mutation). Given that it is simply a matter of independent assortment, with each sibling having a 50% chance of getting one haplotype each from their mother and father, multiply the chances together to get 25%. |
|
|
Which of the following infections is suggestive of a terminal complement deficiency?
mucocutaneous candidiasis recurrent encapsulated organism infections staphylococcal infections recurrent bacterial upper respiratory infections persistent Giardia infection |
RECURRENT ENCAPSULATED ORGANISM INFECTIONS.
Each of the presented choices is suggestive of a particular deficiency. Mucocutaneous candidiasis suggests defective T cells, Staph infections suggest a possible problem with phagocytes, bacterial URIs and giardiasis - a B cell failure. |
|
|
Which of the following is the preferred methodology for assessing HLA haploptype?
microlymphocytotoxicity assay mixed lymphocyte culture assay direct PCR DNA testing serology panel antibody detection direct antibody screening with control antibodies |
DIRECT PCR DNA TESTING.
Nowadays, many of the laborious assays previously utilized to determine HLA haplotypes, such as the options presented in choices A and B, have been replaced by DNA-based assays, such as PCR. Direct DNA analysis is faster, easier, and more precise, allowing for determination of split antigens and other antigens that were not detected by old techniques. |
|
|
How many HLA loci are usually matched for transplantation?
one two three four five |
THREE.
Of course the more the better, but for major antigens a 3 locus (HLA-A, HLA-B, and HLA-DR) match is considered good. In addition in vitro crossmatch is still required because DNA testing does not address antibodies or minor HLA antigens, which could appear to match, but still be incompatible. |
|
|
What does a poor response to S. pneumoniae vaccination potentially indicate?
B cell defect T cell defect B or T cell defect B and T cell defect complement deficiency |
B CELL DEFECT.
Encapsulated organisms, such as S. pneumoniae and N. meningitidis with carbohydrate capsule antigens are opsonized and cleared through the spleen. Therefore, poor response to pneumococcal vaccine is a fairly specific assay for B cell defects. |
|
|
What is the most common specific antibody deficiency?
IgA IgD IgE IgG IgM |
IGA.
Overall, IgA deficiency is the most common. IgG deficiency can be hidden if only a selected subtype is affected. |
|
|
Which of the following scenarios is the most appropriate use of RAST testing?
screening for allergens, especially inhaled ones identifying a specific allergen, especially an inhaled one confirmation of hereditary angioedema confirmation of chronic urticaria diagnosis of systemic mastocytosis |
IDENTIFYING A SPECIFIC ANTIGEN, ESPECIALLY AN INHALED ONE.
In order to perform RAST testing, patient serum is added to a specific antigen pre-complexed with solid phase to which radiolabeled anti-IgE is added. If patient serum has an antigen-specific IgE, then the result would be positive. Since specific antigens are required, it is a poor screening test. The last three choices offered are all independent of IgE, so RAST testing would not be helpful. |
|
|
All of the following modalities are examples of tests of T cell function, except:
delayed-type hypersensitivity skin testing enumeration from peripheral blood smear flow cytometric CD3+ analysis nitroblue tetrazolium assay phytohemagglutinin proliferation assay |
NITROBLUE TETRAZOLIUM ASSAY.
The NBT test provides an assessment of oxidative burst, a function of macrophages and neutrophils. The rest of the tests all provide some indication of T cell number or function. Remember that 3/4 of circulating lymphocytes are usually T cells, so that a peripheral smear usually gives a rough indication of T cell number. |
|
|
All of the following flow cytometric marker patterns are associated with NK cells, except:
CD3- CD4+ CD16+ CD56+ CD57+ |
B. CD4+.
NK cells in their function as mediating cellular immunity do not express CD4, a marker more associated with helper T cells, which modulate B cell and humoral immunity. |
|
|
What is the expected nitroblue tetrazolium assay reaction in patients with chronic granulomatous disease?
red color (non-reactive) <10% f- <10% f+ >50% purple <10% yellow |
C. <10% F+.
The NBT test of oxidative burst assays the catalysis from a yellow color to a purple/blue. A positive test is therefore purple, a pattern referred to as f+ (formazan precipitate positive). Normal neutrophils will be nearly all positive. In chronic granulomatous disease, usually there is less than 10% positivity. |
|
|
Which complement factor levels are assayed for defects in the alternate pathway of activation?
C1q C3 C4 C5 C9 |
C3.
Low level C3 activation is required in order to kick off the alternate pathway in direct response to lipopolysaccharide. For that reason, it is a sensitive, though non-specific, marker for defects in the alternate pathway. An overall screening assay is CH50, which is a functional assay. |
|
|
All of the following are associated with B-cell defects, except:
recurrent bacterial sinopulmonary infections resistant Giardia infections presentation after 6 months of age recurrent staph and strep infections opportunistic viral infections |
OPPORTUNISTIC VIRAL INFECTION.
For the most part, viral and fungal infections are dealt with by cellular immunity and T cells. B cell deficiencies usually present after a few months due to the protection afforded by maternal IgG. Opportunistic recurrent infections with encapsulated bacterial organisms tend to be a major problem. |
|
|
What is the most common presentation of Bruton X-linked agammaglobulinemia?
recurrent pneumonia in middle-aged women persistent sepsis in elderly men resistant upper respiratory tract infections in adolescents meningitis in a neonate chronic diarrhea in a young boy |
CHRONIC DIARRHEA IN A YOUNG BOY.
Most often, GI disease with Giardia is the earliest presentation of Bruton agammaglobulinemia. The characteristic histological finding on GI biopsy is the lack of plasma cells in the interstitium of the mucosa. |
|
|
What is the most commonly inherited immunodeficiency?
Bruton X-linked agammaglobulinemia common variable immunodeficiency selective IgA deficiency Job syndrome severe combined immunodeficiency |
SELECTIVE IGA DEFICIENCY.
There is a very high prevalence (1/700) of IgA deficiency that usually presents with recurrent respiratory and gastrointestinal infections. Patients with IgA deficiency can develop an anti-IgA antibody, which can cause anaphylactic reactions when exposed to IgA-containing products. An important blood bank pearl for IgA deficiency is that patients should get washed products (to remove the IgA against which they could react) or IgA-deficient products. |
|
|
All of the following are associated with diGeorge syndrome, except:
autosomal recessive inheritance pattern defective T cell function hypocalcemia deletion of chromosome 22q11.2 depletion of lymph node paracortical areas |
AUTOSOMAL RECESSIVE INHERITANCE PATTERN.
Velocardiofacial, or diGeorge syndrome, is due to third and fourth pharyngeal pouch fusion and presents with neck, heart, and facial dysfunction/dysmorphia. Because of the neck issues, both the thymus and parathyroids are hypoplastic, leading clinically to T cell defects and hypocalcemia, respectively. The syndrome is due to a sporadic rather than inherited deletion of chromosome 22q11.2. |
|
|
What is the most frequent cause of severe combined immunodeficiency?
autosomal recessive Jak3 deficiency autosomal recessive defect in adenosine deaminase autosomal recessive purine nucleosidase phosphorylase deficiency X-linked recessive defect in IL-2 receptor autosomal recessive CD3 deficiency |
X-LINKED RECESSIVE DEFECT IN IL-2 RECEPTOR.
All of the choices presented are causes of SCID. However, the defects in the X-linked gene, IL-2 receptor, account for the majority of cases with ADA deficiency a close second. All lead to severe defects in both humoral (B cell) and cellular (T cell) responses. |
|
|
The presenting symptoms/signs of Wiskott-Aldrich syndrome include all of the following, except:
microcytic anemia eczema small, uniform platelets thrombocytopenia immunodeficiency |
MICROCYTIC ANEMIA.
The X-linked disorder, Wiskott-Aldrich syndrome (WAS), is characterized by all of the signs and symptoms presented in the question, with the exception of anemia. The WAS gene product, WASP, has been implicated in the disorder. WAS is also one of the causes of platelet dense granule deficiency and bleeding. |
|
|
Which of the following disorders is characterized by cerebellar ataxia, telangiectasias, and recurrent infections?
Duncan disease ataxia telangiectasia chronic granulomatous disease Bruton agammaglobulinemia chronic mucocutaneous candidiasis |
ATAXIA TELANGIECTASIA.
Sometimes the obvious answer is correct! A defect in the DNA mismatch repair gene, ATM leads to a combined T cell and B cell disorder with ataxia and vascular telangiectasias. Since the defect in humoral immunity is primarily IgA, there is usually a history of sinopulmonary infections. There is also a very high (~40%) risk of malignancy, especially hematolymphoid. |
|
|
Duncan disease usually presents as a severe hemophagocytic response to this virus:
HIV HHV8 HHV6 HTLV-I EBV |
EBV.
There are several less severe prodromal signs associated with Duncan disease, but it is the intense response to EBV that defines the disorder. In addition, there are usually subsequent B cell lymphoid malignancies and hepatic failure |
|
|
All of the following are associated with diGeorge syndrome, except:
autosomal recessive inheritance pattern defective T cell function hypocalcemia deletion of chromosome 22q11.2 depletion of lymph node paracortical areas |
AUTOSOMAL RECESSIVE INHERITANCE PATTERN.
Velocardiofacial, or diGeorge syndrome, is due to third and fourth pharyngeal pouch fusion and presents with neck, heart, and facial dysfunction/dysmorphia. Because of the neck issues, both the thymus and parathyroids are hypoplastic, leading clinically to T cell defects and hypocalcemia, respectively. The syndrome is due to a sporadic rather than inherited deletion of chromosome 22q11.2. |
|
|
What is the most frequent cause of severe combined immunodeficiency?
autosomal recessive Jak3 deficiency autosomal recessive defect in adenosine deaminase autosomal recessive purine nucleosidase phosphorylase deficiency X-linked recessive defect in IL-2 receptor autosomal recessive CD3 deficiency |
X-LINKED RECESSIVE DEFECT IN IL-2 RECEPTOR.
All of the choices presented are causes of SCID. However, the defects in the X-linked gene, IL-2 receptor, account for the majority of cases with ADA deficiency a close second. All lead to severe defects in both humoral (B cell) and cellular (T cell) responses. |
|
|
The presenting symptoms/signs of Wiskott-Aldrich syndrome include all of the following, except:
microcytic anemia eczema small, uniform platelets thrombocytopenia immunodeficiency |
MICROCYTIC ANEMIA.
The X-linked disorder, Wiskott-Aldrich syndrome (WAS), is characterized by all of the signs and symptoms presented in the question, with the exception of anemia. The WAS gene product, WASP, has been implicated in the disorder. WAS is also one of the causes of platelet dense granule deficiency and bleeding. |
|
|
Which of the following disorders is characterized by cerebellar ataxia, telangiectasias, and recurrent infections?
Duncan disease ataxia telangiectasia chronic granulomatous disease Bruton agammaglobulinemia chronic mucocutaneous candidiasis |
ATAXIA TELANGIECTASIA.
Sometimes the obvious answer is correct! A defect in the DNA mismatch repair gene, ATM leads to a combined T cell and B cell disorder with ataxia and vascular telangiectasias. Since the defect in humoral immunity is primarily IgA, there is usually a history of sinopulmonary infections. There is also a very high (~40%) risk of malignancy, especially hematolymphoid. |
|
|
Duncan disease usually presents as a severe hemophagocytic response to this virus:
HIV HHV8 HHV6 HTLV-I EBV |
EBV.
There are several less severe prodromal signs associated with Duncan disease, but it is the intense response to EBV that defines the disorder. In addition, there are usually subsequent B cell lymphoid malignancies and hepatic failure |
|
|
Which of the following is a suitable screening test for chronic granulomatous disease?
nitroblue tetrazolium test NAD-coupled oxidation catalase-positive bacterial provocative infection sweat chloride test Benedict's reagent assay |
NITROBLUE TETRAZOLIUM TEST.
Chronic granulomatous disease is due to defects in oxidative burst microbial killing, secondary to a defect in NADPH oxidase. As a result, catalase positive organisms tend to cause severe granulomatous infections due to the immune system's inability to remove the organisms. Nitroblue tetrazolium is a direct assay of the respiratory burst (see question 26 for more). |
|
|
Which of the following treatments can clear the Dohle bodies seen in May-Hegglin anomaly?
DNase RNase iron chelation calcium phosphate precipitation sodium dithionate treatment |
RNASE.
Dohle bodies are large, pale blue inclusions composed of rough endoplasmic reticulum, and are seen in granulocytes and monocytes of the May-Hegglin anomaly. Treatment of cells with ribonuclease can destroy these bodies. Remember that rough endoplasmic reticulum contains numerous ribosomes, which are mostly composed of ribosomal RNA. |
|
|
Which of the following is/are associated with homozygous Pelger-Huet anomaly?
Dohle bodies pince-nez cells Jordan anomaly Stodtmeister cells giant platelets |
STODTMEISTER CELLS.
Heterozygous Pelger-Huet is associated with the more familiar bilobed, or pince-nez, nuclei found in segmented neutrophils. With a homozygous case of Pelger-Huet, monolobated, or Stodtmeister, cells are often seen. |
|
|
What type of disorder usually follows defects in components of the classical pathway of complement activation?
recurrent gram-positive infections hereditary angioedema hematolymphoid neoplasms autoimmune disease recurrent systemic encapsulated organism infections |
AUTOIMMUNE DISEASE.
The vast set of disorders associated with deficiencies of complement proteins reflects the multifaceted role of complement in the body. All of the choices presented (with the exception of hematolymphoid neoplasms) are due to defects in various complement proteins and activation pathways. Recurrent gram positive infections are seen with deficiency of C2 and C3, hereditary angioedema is due to deficiency of C1q esterase inhibitor, autoimmune disease is due to defects in the classical pathway (C1q, C2, and C4) and systemic encapsulated organism infections occur due to deficiencies of membrane attack complex (MAC) components (C5-C9). |
|
|
Which of the following cell types is used most commonly to screen for antinuclear antibody pattern staining?
Vero E6 Hep-2 HeLa human diploid fibroblasts primary monkey kidney |
HEP-2.
Diluted patient serum is added to Hep-2 cells in culture followed by detection with fluorescently-labeled anti-human globulin and a nuclear (DNA) counterstain. From this assay, the classic staining patterns are interpreted. |
|
|
Which of the following staining patterns is most consistent with CREST syndrome?
speckled with mitoses nucleolar with mitoses cytoplasmic centromeric with mitoses homogeneous with mitoses |
D. CENTROMERIC WITH MITOSES.
There's no way around it. The staining patterns are classic, so they have to be memorized. At least centromeric and CREST begin with the same letter. Table 6.3 provides the most common antibody/clinical presentations, including the antinuclear antibodies. It would be worthwhile to spend some time incorporating the chart into your mind! |
|
|
Incubate patient serum (diluted 1:40) with HEp-2 cells, add fluorescein-labeled anti-human globulin (AHG) and counterstain.
Examine for presence and pattern of fluorescence. Homogenous (mitoses+) = ?? Rim (mitoses+) = ?? |
Incubate patient serum (diluted 1:40) with HEp-2 cells, add fluorescein-labeled anti-human globulin (AHG) and counterstain.
Examine for presence and pattern of fluorescence. Homogenous (mitoses+) = dsDNA, ssDNA, histone Rim (mitoses+) = dsDNA |
|
|
Incubate patient serum (diluted 1:40) with HEp-2 cells, add fluorescein-labeled anti-human globulin (AHG) and counterstain.
Examine for presence and pattern of fluorescence. Speckled (mitoses−) = ?? Nucleolar (mitoses−) = ?? Centromere (mitoses+ in centromeric pattern) = ?? Cytoplasmic = ?? |
Incubate patient serum (diluted 1:40) with HEp-2 cells, add fluorescein-labeled anti-human globulin (AHG) and counterstain.
Examine for presence and pattern of fluorescence. Speckled (mitoses−) = Sm, RNP, Scl70, SS-B Nucleolar (mitoses−) = scleroderma Centromere (mitoses+ in centromeric pattern) = CREST Cytoplasmic = AMA, ASMA, etc |
|
|
Screening for antibodies to cytoplasmic constituents
Incubate patient serum (diluted 1:40) with cryostat sections of tissue ‘sandwich’ consisting of rat liver, kidney, and stomach (fundic mucosa & smooth muscle). Add fluorescein-labeled anti-human globulin (AHG). Add counterstain |
Examine for fluorescence
AMA+ in gastric parietal cells, renal tubular cells, and hepatocytes ASMA+ in gastric smooth muscle and renal parenchymal arteries APA+ in gastric parietal cells Anti-LKM+ in cytoplasm of hepatocytes and renal tubular cells Titer all positives |
|
|
What is the use of the microorganism Crithidia luciliae in the screening of antibodies?
it is a positive source of many antibodies it contains giant mitochondria that stain with anti-dsDNA antibodies it is used to produce recombinant proteins that are used as antigens in many tests it effectively blocks interfering antibodies used in the “Crithidia interference assay” to selectively identify most autoantibodies |
IT CONTAINS GIANT MITOCHONDRIA THAT STAIN WITH ANTI-DSDNA ANTIBODIES.
Either the Crithidia test or an ELISA test can be used to screen for anti-dsDNA antibodies. Given the novelty of the Crithidia test, it tends to be asked about more often than the ELISA. |
|
|
What is the effect of age on the detection of anti-nuclear antibody?
decreased false positive rate increased false positive rate decreased false negative rate increased false negative rate no change in the rate of detection |
INCREASED FALSE POSITIVE RATE.
With age, there is decreased test specificity manifested as an increased false positive rate. In addition, the prevalence of autoimmune disease increases with age, which means that, for the positive predictive value of the test to increase with prevalence, the number of true positives must increase more than false positives, relatively….quite confusing. Bottom line - both true and false positive rates of the ANA assay increase with age, true positive a little more than false positive. |
|
|
Increases in the titer of which of the following antibodies can be used to predict lupus flares?
anti-histone anti-Smith anti-SSA anti-dsDNA anti-RNP |
D. ANTI-DSDNA.
All of the choices are associated with lupus: histone, dsDNA, and RNP mostly with drug-induced lupus, anti-Smith with systemic lupus, and SSA both with lupus and Sjogren syndrome. Anti-RNP is also associated with mixed connective tissue disease. Of all of the antibodies, anti-dsDNA is the most sensitive to changes in lupus status, while anti-Smith is the most specific for SLE. |
|
|
All of the following conditions are associated with anti-mitochondrial antibodies, except:
primary biliary cirrhosis syphilis ulcerative colitis collagen vascular disease cardiomyopathy |
ULCERATIVE COLITIS.
There are many antigens in mitochondria against which antibodies can be raised. Different anti-mitochondrial membrane antibodies are associated with different diseases - M2 with primary biliary cirrhosis, M1 with syphilis, M5 with collagen vascular disease, M7 with cardiomyopathy. Ulcerative colitis is often seen with primary sclerosing cholangitis (more often PSC is seen with ulcerative colitis than the converse) and is associated with p-ANCA. |
|
|
What antigen is c-ANCA responsive to?
proteinase 3 lipoprotein lipase myeloperoxidase gluteraldehyde dehydrogenase pyruvate kinase |
PROTEINASE 3.
So called because of its cytoplasmic staining pattern, c-ANCA is directed against the cytoplasmic protein, c-ANCA. It is most specific for Wegener granulomatosis. p-ANCA, or perinuclear ANCA, is directed against the predominant perinuclear myeloperoxidase antigen, and is less specific than c-ANCA. |
|
|
How is a “lupus erythematosus” cell defined?
a phagocytic cell with an engulfed denatured nucleus a dessicated cell surrounded by a wreath of T-cells a binucleate giant cell with a prominent nucleolus a ghost cell after incubation with patient serum an opsonized clumped red blood cell in the peripheral smear |
A PHAGOCYTIC CELL WITH AN ENGULFED DENATURED NUCLEUS.
The LE cell assay has a high sensitivity for lupus, nearly 70%. The assay is performed by agitating tissue or body fluids in a tube and then looking for the characteristic LE cell - a phagocytic cell with an engulfed denatured nucleus. |
|
|
What feature do all conditions associated with increased angiotensin-converting enzyme have in common?
all form granulomas all are in the kidney all present with cholestasis all affect the seminiferous tubules all have a characteristic very high white blood cell count |
ALL FORM GRANULOMAS.
While all granulomatous diseases are not associated with increased ACE, most disorders associated with increased ACE are granulomatous in nature - sarcoidosis, leprosy, primary biliary cirrhosis, and Gaucher disease. In the case of sarcoidosis, ACE levels are useful in diagnosing a flareup of disease. |
|
|
All of the following are associated with HLA-DR3, except:
insulin-dependent diabetes mellitus ankylosing spondylitis systemic lupus erythematosus myasthenia gravis celiac sprue |
ANKYLOSING SPONDYLITIS.
HLA haplotypes are more strongly associated with disease than would be expected by chance. HLA-DR3 is associated with all of the choices except ankylosing spondylitis, which is associated with HLA-B27. Several other HLA haplotypes, especially HLA-DR ones are associated with a number of other autoimmune disorders. |
|
|
Which of the following drugs is/are associated with drug-induced lupus?
hydralazine procainamide isoniazid A & B A, B, C |
E. A, B, C.
All are associated with drug-induced lupus, which in turn is highly associated with anti-dsDNA and anti-histone antibodies. |
|
|
What type of hypersensitivity is responsible for a positive reaction to a tuberculin skin test?
Type I Type II Type III Type IV Type V |
TYPE IV.
First of all, there is no Type V. The hypersensitivity reactions can be remembered with the mnemonic “ACID.” Type I is Antigen-Antibody-mediated and immediate, like in anaphylaxis. Type II is Cell-mediated when antibody binds antigen to activate cellular toxicity, like in myasthenia gravis. Type III is Immune complex-mediated when antibody-antigen-complement complexes are deposited, like in SLE. Finally, Type IV is Delayed, where antibodies bind antigen and active cellular immunity, like in a positive skin tuberculin test. |
|
|
All of the following are associated with celiac sprue, except:
unexplained short stature and iron-deficiency anemia HLA-DQ2/8 IgA deficiency dermatitis herpetiformis increased intraepithelial neutrophils in the duodenum |
E. INCREASED INTRAEPITHELIAL NEUTROPHILS IN THE DUODENUM.
Often, the earliest histological manifestation of sprue will be increased intraepithelial lymphocytes. This change often precedes the characteristic villous blunting and crypt elongation. There are many other conditions and findings associated with sprue, including diabetes mellitus, dermatitis herpetiformis, cystic fibrosis, enteropathy-associated T cell lymphoma, arthritis, and malnutrition. |
|
|
All of the following are useful in the diagnosis of autoimmune thyroiditis, except:
anti-microsomal antibodies TSH receptor antibodies anti-thyroglobulin antibodies thyroglobulin levels long-acting thyroid stimulating antibodies |
THYROGLOBULIN LEVELS.
For the most part, the diagnosis of autoimmune thyroiditis depends on demonstration of some anti-thyroid antibodies, whether it's Hashimoto, which is associated with anti-thyroglobulin and anti-microsomal antibodies, or Graves, which is associated with LATS or TSH receptor antibodies. |
|
|
What's the most specific serum assay for the diagnosis of lymphoplasmacytic sclerosing pancreatitis?
carbonic anhydrase antibodies IgG4 levels anti-smooth muscle antibodies angiotensin-converting enzyme levels anti-liver/kidney microsomal antibody |
IGG4 LEVELS.
The majority of cases of LPSP present with an increased serum IgG4 fraction, which is often identified by increased IgG4-expressing plasma cells within the lesion. Anti-carbonic anhydrase, though less sensitive, is also associated with LPSP. |
|
|
All of the following antibodies have been associated with autoimmune hepatitis, except:
ANA anti-SLA/LP anti-smooth muscle antibody c-ANCA anti-LKM1 |
C-ANCA.
Anti-smooth muscle antibody is the most commonly associated antibody with autoimmune hepatitis, but it is important to note that there are several other antibodies also commonly associated with autoimmune hepatitis. In fact, there may be several subgroups of the disease with different antibodies associated with each. |
|
|
What is the gold standard of diagnosis for giant cell arteritis?
confirmation of concurrent polymyalgia rheumatica temporal artery biopsy increased serum anti-CRP antibody aortic biopsy increased erythrocyte sedimentation rate |
TEMPORAL ARTERY BIOPSY.
Giant cell arteritis was previously called temporal arteritis, but due to its more widespread presentation, it was renamed. However, the temporal artery is still the most commonly affected with headache, and visual changes are the most common presentation. There are often increased ESR and CRP levels, but the temporal artery biopsy is the gold standard. An aortic biopsy? Are you kidding? |
|
|
Which of the following antibodies is associated with poor prognosis, myositis, high frequency of cardiac manifestations, and HLA-DR5?
anti-titin anti-SRP anti-Mi-2 anti-SS-A anti-MuSK |
ANTI-SRP.
The myositis diseases are an assortment of anti-muscle autoimmune disorders with various antibodies and presentations. In addition to being de novo autoimmune disorders, they are also frequently seen as paraneoplastic conditions or associated with other autoimmune disorders. The anti-SRP antibody has the worst prognosis and is most commonly associated with polymyositis. |
|
|
Which of the following antibodies is most associated with the majority of types of myasthenia gravis?
anti-AChR anti-MuSk anti-titin anti-synaptophysin anti-cholinesterase |
ANTI-ACHR.
MG presents with the characteristic progressive muscle weakness/fatigue that is relieved by cholinesterase inhibitors (the basis of the Tensilon test). There are several subtypes of MG, including those cases associated with thymoma, each associated with different antibodies. The anti-AChR antibodies tend to be found in almost all types of MG with the exception of the so-called seronegative cases, which are more commonly associated with anti-MuSK antibodies. |
|
|
Which of the following assays best reflects mast cell degranulation?
serum total tryptase serum mature tryptase serum histamine urinary histamine urinary HIAA |
SERUM MATURE TRYPTASE.
Both tryptase and histamine are released into the serum in cases of anaphylaxis. Histamine is less specific than tryptase and can be also seen in scombrodic fish poisoning. Both total and mature tryptase are produced by mast cells with total representing the overall amount of mast cells and mature more a reflection of mast cell degranulation. |
|
|
|