Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
61 Cards in this Set
- Front
- Back
|
Physiology of Haemostasis
Haemostatis |
is a process that halts bleeding following vascular injury
– Involves complex relationship between substances promoting clot formation (platelets, coagulation cascade), inhibit coagulation, and dissolve formed clot |
|
|
describe process of Clot Formation
|
Vascular injury (eg. Ruptured plaque, trauma,) results in endothelial damage
• Vasconstriction occurs to limit blood flow to area • Subendothelial structures (eg. Collagen, basement membrane, fibronectin) become exposed and stimulate platelets. • Platelets adhere to site of damage (stimulated initially by fibrinogen) using circulating von Willebrand factor as a ligand between subendothelium and glycoprotein Ib receptors on platelets • Platelets de-granulate, releasing ADP, TXA2, Platelet Factor 3, Platelet Factor 4, Prostacyclin, Serotonin, phospholipids, and lipoproteins. These substances play various roles including platelet aggregation and stimulation, vasoconstriction, vasodilation, inflammatory responses, etc Platelets change shape and express receptors for Factors V and VIII and leucocytes on surface • Coagulation cascade activated with available clotting factors adhering to surface of platelet • Activation of clotting cascade makes Thrombin and Fibrin available • Platelet aggregation and cross-linking of Fibrin strands completes clot formation, stabilises clot, and theoretically stops bleeding • White blood cells attracted to area, bind to surface receptors, set up inflammatory responses and initiates healing process • Activation of the coagulation cascade also activates the fibrinolytic system and other inhibitory substances which serve to counter balance, and hence help to prevent over coagulation. • It can be seen that platelets form a critical component of the clotting cascade |
|
|
Clotting Cascade
|
The coagulation system has long been termed a cascade as each activated
factor serves as a major catalyst for the next reaction. Thus small amounts of early activated factors results in a large amount of thrombin and fibrin • Historically, the cascade has been divided into two distinct arms – Intrinsic Pathway activated by physical/chemical injury to vascular endothelium exposing subendothelial substances including Collagen – Extrinsic Pathway activated by tissue factors released from damaged cells – Common Pathway – Intrinsic and Extrinsic pathways meet at the activation of Factor X. |
|
|
cascade flow chart
|
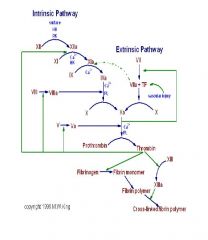
|
|
|
Intrinsic Pathway - stimulas and sequence of events
|
Stimulus from damage to vascular endothelium eg. Plaque formation, rupture
• Sequence of events is – Inactive Factor XII to active XIIa – Platelets activated – Factor XIIa is a protease which enzymatically activates Factor XI to XIa in presence of High Molecular Weight Kininogen & Prekallekrein. Factor XIIa also promotes conversion of Prekallekrein to Kinogen (thus promoting the cascade) – Factor XIa converts Factor IX to IXa (in the presence of Ca) and leads to the release of Bradykinin from HMWK – Factor IXa converts Factor X to active Xa in the presence of platelet membrane lipoprotein, Factor VIII and Calcium; this reaction is greatly accelerated by Factor VIIIa – Factor VIII activation is initially stimulated by small quantities of Thrombin; as concentration of Thrombin increases VIIIa is deactivated (serves as a counterbalance) – Factor Xa is part of the common pathway, and acts as a protease to convert inactive molecular prothrombin (Factor II) to active Thrombin in the presence of Factor V – Thrombin then cleaves Fibrinogen to Fibrin, which polymerises to form fibrin strands. |
|
|
Intrinsic Pathway - what is a measured by, what is not altered, what factor defiencies lead to haemophilia
|
Intrinsic pathway feeds into the final common pathway.
• Deficiencies of Factors VIII or IX lead to Haemophilia • Measured by Activated Partial Thromboplastin Time (aPTT) • Commonly used for evaluation of heparin therapy. • Note Low Molecular Weight Heparins do not generally alter aPTT as effects of these largely on Factor X |
|
|
Extrinsic Pathway - stages
|
Initiated at the site of injury by release of tissue factors
(tissue thromboplastin /Factor III) • Tissue Factor combines with Factor VII in presence of Calcium to produce a complex which activates VII • Factor VIIa complex plus Calcium and phospholipid activates Factor X to Xa • The ability of factor Xa to activate factor VII creates a link between the intrinsic and extrinsic pathways. • An additional link between the two pathways exists through the ability of tissue factor and factor VIIa to activate factor IX. • The Coagulation Cascade proceeds down the Common Pathway to produce a clot • A major mechanism for the inhibition of the extrinsic pathway occurs at the tissue factor--factor VIIa--Ca2+--Xa complex. The protein, lipoprotein associated coagulation inhibitor, LACI (also known as tissue factor pathway inhibitor, TFPI) specifically binds to this complex. TFPI binds to factor Xa and and to factor VIIa (only in the presence of factor Xa.) |
|
|
Extrinsic Pathway - what is it measure by - and what is this test used for
|
The Extrinsic pathway is measured by the Prothrombin Time (PT)
• Prothrombin Ratio used for evaluation of Warfarin therapy • INR is standardised PR against an international control |
|
|
Common Pathway - how does it start and whats the process
|
Both the Extrinsic and Intrinsic pathways activate Factor X in the
final Common Pathway • Factor X activation allows the eventual formation of Thrombin • Prothrombinase + Phospholipid (eg PF3) is a complex that forms when Factors Xa and V, platelet phospholipid, and Calcium combine to become an enzyme like substance. • This complex converts Prothrombin to Thrombin • Thrombin is a potent procoagulant; it catalyses activation of V and VIII through positive feedback, resulting in more conversion. • Thrombin also stimulates platelets, cleaves prothrombin to more thrombin, cleaves fibrinogen to form stable insoluble fibrin plug • Thrombin also has inhibitory effects at high concentrations by deactivating Factor VIIIa • Note that the cascade accelerates with each step, with the amount of product formed increasing logarithmically |
|
|
what are Inhibitors of Coagulation
|
Protein S
Protein C Tissue Factor Pathway Inhibitor (TFPI) Antithrombin III (AT, AT III) Thrombomodulin The Fibrinolytic System |
|
|
another cascade chart
|
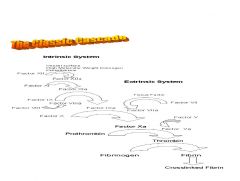
|
|
|
Inhibitors of Coagulation Protein S, Protein C, Tissue Factor Pathway Inhibitor (TFPI)
|
Numerous mechanisms are present to limit coagulation, and
balance the coagulation cascade including • Protein C – naturally occurring, Vitamin K dependent anticoagulant – Activated by high concentrations of Thrombin – Destroys Factors V and VIII • Protein S – Vitamin K dependent anticoagulant – Deactivates Va and VIIIa • Tissue Factor Pathway Inhibitor (TFPI) – protein that mediates the feedback inhibition of the Tissue Factor-Factor VIIa complex – decreases activation of both Factor IX and X. – Small amounts of Factor Xa are required in order for TFPI to achieve its inhibition of Factor VIIa-Tissue Factor complex, – thus Xa inhibits its own production – (note Heparin increases TFPI activity by 2-4fold) |
|
|
Inhibitors of Coagulation
Antithrombin III (AT, AT III) Thrombomodulin |
Antithrombin III (AT, AT III)
– protein synthesized by liver and endothelial cells which binds and directly inactivates thrombin and the other serine proteases (Factors IXa, Xa., and XIa). – reaction between the serine proteases and AT is relatively slow. – The serine proteases still have time to generate thrombin and fibrin before becoming inactivated. – heparin catalyses reaction which becomes virtually instantaneous resulting in the immediate blockage of fibrin formation. • Thrombomodulin – endothelial cell receptor which binds thrombin – forms a complex, which changes the conformation of the thrombin – altered thrombin molecule now readily activates Protein C and loses is platelet activating and protease activities. – Therefore, the binding of thrombomodulin to thrombin converts thrombin from a tremendously potent procoagulant into an anticoagulant. – normal endothelial cells produce thrombomodulin which binds any circulating thrombin, thus preventing clot formation in undamaged vessels. |
|
|
Inhibitors of Coagulation
• The Fibrinolytic System |
The Fibrinolytic System
– keeps clot formation in check by degrading the fibrin strands. – Plasminogen is an inactive protein made in endothelial cells, liver cells, and eosinophils – Fibrin in clots binds circulating plasminogen – Plasminogen is activated to plasmin by an enzyme (tissue plasminogen activator, tPA) in vascular endothelial cells – Release of tPA is stimulated by thrombin – Plasmin degrades fibrin strands, preventing the build-up of excess clot. – Fibrin Degradation Products (FDPs) generated from breakdown – Plasmin released into circulation by clot dissolution is rapidly deactivated by enzymes – Interference with Fibrinolytic system • Fibrinolytic drugs eg Streptokinse, Urokinase, tPA,tPA derivatives • Inhibitors of fibrinlysis eg Tranexamic Acid, Aminocaproic acid, Aprotinin |
|
|
diagramatic representation of regulation of cascade
|
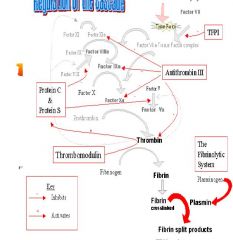
|
|
|
Laboratory Test of Coagulation system - considerations
|
Laboratory tests critical in the diagnosis, monitoring, and treatment
of coagulation disorders. • used appropriately, they can yield a great deal of information and can often localize a disease process to a certain part of the cascade. • tests are extremely helpful in the monitoring of anticoagulant therapy. • tests reach their highest specificity and sensitivity only when the clinical picture is also considered • most common laboratory evaluations used are the activated prothrombin time (PT) and activated partial thromboplastin time (aPTT). Other tests such as thrombin time and bleeding time are also extremely valuable, but often underused. |
|
|
Protthrombiin Tiime (PT) - purpose and method
|
Screening test for Extrinsic Pathway
Purpose – Screening test to identify acquired or inherited deficiencies in the activities of factors VII, X, V, prothrombin and fibrinogen. eg liver disease, Vitamin K deficiencies, DIC – Monitor oral anticoagulant therapy with warfarin, which decreases the activity of VII, IX, X and prothrombin Method – Collect blood from the patient into a citrated tube – Take the collected blood to the laboratory – Centrifuge to isolate the plasma – Take small sample of the plasma ~0.1 ml. – Add calcium – Add Thromboplastins - which are preparations of tissue factor and phospholipids via genetic engineering. – The time that elapses until clot forms is the activated Prothrombin Time (PT). (Normal 11-13 sec) |
|
|
Protthrombiin Tiime (PT)
Interpretation - and abnormalitie causes |
PT shorter than the reference range is not associated with any clinical
condition and is usually related to improper procedure. – Prolongation > 2 seconds is abnormal – Prolongation of the PT occurs when there is: • Increased hematocrit, leading to a false positive in PT prolongation. • Abnormalities in thrombin generation • Liver disease • Vitamin K deficiency • Disseminated intravascular coagulation • Nephrotic Syndrome • Treatment with certain antibiotics, chemotherapeutics, or antithrombotic drugs. – Note: The PT will increase shortly after a bolus dose of heparin. – INR has generally taken over as test of choice for monitoring of oral anticoagulants because of standardisation of the test |
|
|
Parttiiall Thrombopllasttiin Tiime (aPTT) - purpose
|
(Normal 28-33 sec)
Screening test for abnormalities in Intrinsic Pathway triggered by exposure of plasma to negatively charged surfaces • Measures Factors XII, XI, IX, VIII, X, and V, Fibrinogen, Prothrombin, prekalikrein, high molecular weight kininogen Purpose – Screening test to identify acquired or inherited deficiencies in the activities of Factors IX, VIII, and XI. – Screening test for lupus anticoagulants – Monitor heparin anticoagulation – Screening test to assess reduction in the activity of fibrinogen, Factors V and X. However, the PT is more sensitive for these. |
|
|
Parttiiall Thrombopllasttiin Tiime (aPTT) (Normal 28-33 sec)
Method |
Parttiiall Thrombopllasttiin Tiime (aPTT) (Normal 28-33 sec)
Method – Collect blood from the patient into a citrated tube & centrifuge to isolate the plasma – Take a small sample of the plasma ~0.1 ml – Add calcium (catalyst for coagulation) and partial thromboplastinphospholipid source without tissue factor (= the procoagulant) – Add a negatively charged surface such as kaolin (usually), silica, celite, or dextran sulphate to activate coagulation (= surface active agent) – The time that elapses until clot forms is the activated Partial Thromboplastin Time (aPTT). |
|
|
Parttiiall Thrombopllasttiin Tiime (aPTT) (Normal 28-33 sec)
Interpretation |
Parttiiall Thrombopllasttiin Tiime (aPTT) (Normal 28-33 sec)
Interpretation – aPTT shorter than the reference range is usually the result of poor sample handling. – Prolongation of the aPTT occurs when there is: • Factor Deficiency – aPTT sensitive to deficiencies below 30-40% of normal of all clotting factors except VII or XIII • Factor Inhibitor eg lupus anticoagulant and Factor VIII inhibitors. – Possible to distinguish between deficiency and inhibitor by mixing sample with pooled blood. » Deficiency – no change aPTT (deficiency corrected by factors in pooled blood), » Inhibitor – prolonged aPTT (as inhibitor will affect factors in pooled blood) • Anticoagulation with heparin – aim of Heparin treatment is aPTT 1.5 to 2.5 times normal ie 65-80sec • Contamination of sample with heparin |
|
|
Thrombiin Tiime (TT) - purpose and method
|
Purpose
• Screening test for last step in coagulation pathway • Screening test for changes in fibrinogen and fibrin – Eg. hypofibrinogenemia, hyperfibrinogenemia, abnormalities of the fibrinogen molecule, and inhibitors against thrombin or fibrin. • Tests the conversion of fibrinogen to fibrin to cross-linked fibrin. • Monitor anticoagulant therapy with fibrinolytic agents and hirudin.. • When modified (using high concentration of thrombin), the test can be used to measure fibringoen levels. Method • Collect blood from the patient into a citrated tube and centrifuge to isolate the plasma • Take small sample of the plasma ~0.1 ml. • Add bovine thrombin to plasma • The time that elapses until clot forms is the Thrombin Time (TT). |
|
|
Thrombiin Tiime (TT)
Interpretation |
Thrombiin Tiime (TT)
Interpretation • TT shorter than the reference range is not associated with any clinical condition is usually related to improper procedure. • Prolongation of the TT occurs when there is: – Contamination with heparin. TT can be repeated after addition of protamine sulfate and will be normal. – Afibrinogenemia/Hypofibrinogenemia eg. • acquired (DIC, FDP’s, liver disease) and familial – Interference with fibrinopeptide cleavage |
|
|
Intternattiionall Normalliised Rattiio (INR)
|
Intternattiionall Normalliised Rattiio (INR)
(Normall <1..2,, Therapeuttiic 2..0-3..5) • Ratio of Prothrombin Time of test to standard control • Developed due to major differences in thromboplastin reagents used which led to variable results between labs • Manufacturers of reagent supplied with International Sensitivity Index number for each batch of reagent • This is a major test which is used to monitor therapy with oral anticoagulants eg. Warfarin • Aim of Warfarin therapy is to achieve an INR of 2-3 times upper limit of normal (depending on indication) • We must be familiar with this test |
|
|
Platlets - normal range, function
|
Function
– maintain integrity of blood vessels – interact to facilitate blood coagulation • provide specific receptor site for clotting factors • provide necessary phospholipid surface for conversion of prothrombin to thrombin • protection of thrombin from enzyme antithrombin Evaluation of platelets important in assessment of bleeding disorders |
|
|
Plalets
• Production & metabolism |
Plalets
• Production & metabolism – produced in bone marrow as megakaryoctes – megakaryocte maturation & proliferation controlled by megakaryocte colony stimulating factor (Mk-CSF) & thrombopoietin – 2/3 platelets in circulation, 1/3 in spleen – life of platelet 8 - 11 days; young platelets are more functional than old platelets – Spleen site of destruction |
|
|
Plalets
Tests |
Pllattelletts
Tests • Initial testing is by count – automated (most accurate between 50,000 and 500,000) or manual count (may be performed if < 50,000) • Normal Platelet count is 160,000 to 420,000 • Visualisation of platelets may assist in diagnosis – New platelets are large and elongated – Old platelets are small • Abnormalities in platelet counts may be associated with significant changes in coagulation • Megakaryocytes generally only found within bone marrow and can be measured with bone marrow aspiration |
|
|
Pllattelletts - Thrombocyttopeniia
Decrease counts (Thrombocytopenia) |
Pllattelletts - Thrombocyttopeniia
Decrease counts (Thrombocytopenia) • Defined as a platelet count of less than 150,000, although haemostasis approaches normal at levels > 50,000 • Often asymptomatic and of no clinical importance eg. Counts of 100,000 • Becomes a major clinical issue when count <20,000 to 30,000 • Mechanisms include – Increased Destruction or sequestration – Decreased production – Ineffective production – Dilution Causes include • neoplastic diseases • immune processes • infections • metabolic disorders • drugs & chemicals • Spleenomegaly • bone marrow failure eg Aplastic anaemia |
|
|
Pllattelletts - Thrombocyttopeniia
Increased Destruction or sequestration |
Pllattelletts - Thrombocyttopeniia
Increased Destruction or sequestration – Hypersplenism – rarely severe enough to require splenectomy – Thrombotic Thrombocytopenic Purpura (TTP)/Haemolytic Uraemic Syndrome (HUS) • associated with microangiopathic haemolysis; • aetiology often uncertain; includes sepsis eg E.Coli Renal failure prominent in HUS – DIC • essentially always present in acute DIC • Marrow compensation may maintain counts in chronic DIC • Counts return to normal after correction of DIC – Sepsis (especially Gram Negative) • May develop thrombocytopenia in absence of DIC • Endotoxins sequestrate platelets in capillary circulation – Immune Thromboctyopenia • Antibody mediated, although proof of antibodies often difficult eg IgG testing is sensitive but lacks specificity; IgG may not be aetiological agent • Frequently severe with counts <10,000 • May be acute or chronic |
|
|
Pllattelletts - Thrombocyttopeniia
decreased production ineffective production dilutional |
Pllattelletts - Thrombocyttopeniia
Decreased Production • Bone Marrow aspirate reveals decreased megakaryocytes • Causes include – Extensive bone metastes – Acute leukamias, CLL, multiple myeloma, – Aplastic anaemia – Infections especially viral – Drugs – cytotoxic agents, thiazides, Alcohol, immunosuppressants Ineffective Production • Megakaryocytes present in bone marrow but platelet production is defective • Causes include – Megaloblastic anaemias eg B12 and folate deficiency, – Drug induced megaloblastosis Dilutional • Significant volume overload may lower platelet count – generally not significant • Replacement of blood with blood substitutes may lower counts |
|
|
Pllattelletts - Thrombocyttopeniia
Immune Thromboctyopenia |
Pllattelletts - Thrombocyttopeniia
Immune Thromboctyopenia – Differential diagnosis • Primary (Idiopathic Thrombocytopenia Purpura or ITP) • Secondary – Viral infections, – Drugs – numerous drugs – Collagen diseases (eg.SLE), – Lymphoproliferative disorders, – Graves disease, – Immunodeficiency states |
|
|
Pllattelletts - Thrombocyttopeniia
Immune Thromboctyopenia - treatment for acute and chronic |
Pllattelletts - Thrombocyttopeniia
Immune Thromboctyopenia Treatment • Acute immune thrombocytopenia – Identify cause and withdraw (often drugs) – Severe with symptoms – High dose steroids eg. Prednisolone 25- 50mg/d for 10-21 days • Chronic ITP – High dose steroids (25-100mg Prednisolone per day) – Response in 60-70% patients in 3 weeks – Taper steroids when platelets> 100,000 – Consider spleenectomy if unresponsive – Danazol (androgen analogue) 200mg tds/qid and taper as possible – IV Intragam (gamma globulin) trial – expensive, short action, |
|
|
Drug induced Thromboctyopenia - name drugs
|
Drug induced Thromboctyopenia
• Most common of drug induced haematological disorders. • May be due to direct bone marrow toxicity or immune mediated • In direct toxicity will be decreased megakarocytes cf increased megakarocytes in immune mediated due to peripheral destruction of platelets • Large number of drugs have been associated with thrombocytopenia • Cytotoxic agents most common cause, followed by Heparin • Other agents include – Allopurinol, Amphotericin, antiviral agents, Aspirin, – Cephalosporins, Chloramphenicol, – Frusemide, Gold, H2 antagonists, Hydroxychloroquine, – Interferon, Isoniazid, Methotrexate, – Penicillins, Phenytoin, Procainamide, Propylthiouracil, – Quinine/Quinidine, – Rifampicin, – Sodium Valproate, Sulphonamides, – Thiazides, Trimethoprim, etc. |
|
|
Drug induced Thromboctyopenia
Heparin |
Drug induced Thromboctyopenia
Heparin • Occurs in 25% patients treated with Heparin • Not associated with dose • Two distinct types – Early thrombocytopenia • Mild decrease to approx 100,000 • Nil symptoms, reverses rapidly • ? Due to sequestration of platelets – Late thrombocytopenia • Late onset (6-10days) • Known as Heparin Induced Thrombocytopenia syndrome (HITS) • Severe thrombocytopenia with counts < 30,000, associated with bleeding, thrombosis and Heparin resistance, • Likely Immune mediated with antibody to Heparin/platelet complex; antibodies also may attach to endothelial cells on blood vessel walls promoting thrombosis. • Rates reported 2.9-5.4% beef Heparin, 1.1-1.3% pork Heparin • Low molecular weight Heparins have a lower incidence, but may still induce HITS (potential can be evaluated in laboratory by incubation patients platelets with LMWH) • Danaparoid is anticoagulant of choice in patients with HITS or history of HITS |
|
|
Drug induced Thromboctyopenia
thiazides alcohol gold QUININE/QUINIDINE |
Drug induced Thromboctyopenia
GOLD • Incidence 1-3% - may be abrupt and severe or delayed by months • Associated with HLA typing (DR-3&4) • Antibody formed to platelet QUININE/QUINIDINE • Innocent bystander immune mechanism (antibody vs drug destroys platelet) • Low affinity Hapten type reaction most common but antibodies against specific platelet membrane glycoproteins have been identified • Thrombocytopenia may occur with initial exposure (7-10 days treatment needed); • Secondary exposure to even minute amounts will precipitate reaction • Platelet count may afll dramatically with early onset blleding THIAZIDES • Direct bone marrow suppression likely although specific antibodies have been identified ALCOHOL • General marrow suppressant • Rates – hospitalised alcoholics 26%, well alcoholics 3% • Chronic alcohol causes decreased production, decreased survival in circulation, and platelet dysfunction |
|
|
Increased Pllattelletts - Thrombocyttosiis - causes features treatment
|
Significant thrombocytosis may be associated with increased risk of vascular
events Causes include – Myeloproliferative disorders eg. • Polycythemia Rubra vera • Primary Thrombocythemia • Agnogenic nyloid metaplasia • Chronic granulocytic leukaemeia Features – In myeloprolific disorders platelet counts often < 1,000,000 – Platelets morphologically bizarre & functionally abnormal – Bleeding common, especially when platelet count very high – Platelets also tend to clump in microcirculation and may contribute to thrombosis Treatment – Antiplatelet agents considered necessary – Cytotoxic agents may have a role eg Hydroxyurea, alkylating agents |
|
|
Increased Pllattelletts - Thrombocyttosiis
Causes include • Secondary Thrombocytosis |
Increased Pllattelletts - Thrombocyttosiis
Causes include • Secondary Thrombocytosis • Inflammation • Malignancy • Hodgkin’s Disease • Acute bleeding • Post-spleenectomy • Rebound from severe thrombocytopenia • Severe Iron deficiency anaemia Features – Tends to be less severe – Bleeding less likely Treatment – Treatment of underlying cause |
|
|
Anttiitthrombiin III
Review: Antithrombin III (AT): |
Anttiitthrombiin III
Review: Antithrombin III (AT): – Synthesized in liver and endothelial cells – Acts as an anticoagulant by directly binding and inactivating the serine proteases (Factors XIa, IXa, Xa, and Thrombin) – The presence of heparin increases the activity of AT by 104 - 105. – Major action of Heparin is via Antithrombin III – Normal response to Heparin requires at least 40% normal AT III levels – Deficiencies of AT III increases risks of thromboembolic disorders. |
|
|
Anttiitthrombiin III
AT III deficiency: causes |
Anttiitthrombiin III
AT III deficiency: – A Defect in synthesis of AT III • Congenital: Deficiencies of AT III occur in 1 in 2,000 – Type 1 - Classic - There is reduced synthesis of the AT molecule. A gene deletion or frameshift mutation results in a truncated protein which is unstable. – Type 2 - The level of AT produced is normal, but the protein is dysfunctional. – Type 3 - The quantity and quality of AT is normal but it lacks the receptor for heparin; therefore, it cannot be accelerated. • Acquired: Causes include – acute thrombosis (70% patients with significant DVT or PE have decreased ATIII levels pretreatment, which correct with treatment), – DIC, – liver disease (decreased levels but generally not associated with thrombosis) – nephrotic syndrome (levels increase with steroid treatment; ? Direct stimulation or ? Decreased protein loss), – oral contraceptive use. – B. Increased consumption of AT III due to higher levels of serine proteases, which is often seen in: • Disseminated intravascular coagulation (DIC) • Extensive deep venous thrombosis (DVT) • Pulmonary embolus – C. Loss of AT III - i.e. nephrotic syndrome – D. Increased protein catabolism |
|
|
Anttiitthrombiin III
• Clinical features of AT deficiency: |
Anttiitthrombiin III
• Clinical features of AT deficiency: – Increased risk of venous thrombosis and pulmonary embolism. Venous thrombosis occurs most frequently in the deep veins of the lower extremities. – Thrombotic events begin in mid-late teenage years. – Mesenteric veins, inferior vena cava, renal veins are all susceptible. – Cerebral vein thrombosis can occur. – Events occur with AT activity at 40-60% of normal. Homozygosity is fatal in utero. – May be precipitated by provocations such as surgery, trauma, pregnancy, oral contraceptive (OCP) use, or infection. – Arterial thrombotic events are not increased. – True congenital deficiency may commit the patient to life long anticoagulation. |
|
|
Anttiitthrombiin III
• Treatment of AT deficiency: |
Anttiitthrombiin III
• Treatment of AT deficiency: – Will depend on the cause – Anticoagulation with heparin or Warfarin mainstay of therapy – AT III concentrates are available for short term treatment – True congenital deficiency may commit the patient to life long anticoagulation. – Acquired AT III deficiencies may respond to treatment of underlying disorder eg Steroids for Nephrotic Syndrome |
|
|
Protteiin C - characteristics
|
Protteiin C
• Naturally occurring Vitamin K dependent anticoagulant • inhibitor of the procoagulant system • catalyses the proteolysis of Factors VIIIa and Va, and hence inhibit their coagulant actions. • Synthesised in the liver as inactive form and is Protein C is activated when thrombin binds to thrombomodulin. This binding alters the conformation of thrombin to a form that readily activates Protein C. • Protein S acts as a cofactor and increases activity of Protein C • Protein C deficiency occurs in 1 in 200-300 (autosomal dominant), although clinical disease is less common. Presentation is with thromboembolic disease • Testing difficult - technical problems, interference (Heparin, Warfarin, clotting) |
|
|
Protteiin C - clincial features - implications
|
Protteiin C
• Clinical Features – Increased risk thromboembolism, especially DVT and PE – Clotting disorders often present in late teenager years – Thrombosis may occur in cerebral veins, retinal veins, mesenteric veins, renal veins, and the inferior vena cava – Clotting may be further precipitated by surgery, trauma, pregnancy, or oral contraceptive use. – Neonatal purpura fulminans often fatal – Coumadin Skin Necrosis • Implications are – Acute thrombosis best treated with Hepain – life long anticoagulation generally necessary – Warfarin /sc Heparin – increased tendency for clotting with Warfarin initiation – therapy must be commenced with low doses and very gradually increased – increased potential for Warfarin skin necrosis – requires cessation Warfarin and initiation of Heparin • |
|
|
Protteiin S - where is acquired defiency
|
Protteiin S
• Naturally occurring Vitamin K dependent anticoagulant • Co-factor for Protein C - catalyses the proteolysis of Factors VIIIa and Va, and hence inhibit their coagulant actions • In the circulation, Protein S exists in two forms: a free form and a form bound to complement protein C4b. • Congenital deficiency less common than Protein C deficiency • Clinical features of Protein S deficiency are similar to Protein C Deficiency – 50% will have first thrombotic event before age 25 – 44% will have other provocations, while the remaining 56% will have spontaneous thrombosis. • Acquired deficiency – Seen in consumptive processes such as DIC or extensive DVT/PE. – Seen in patients taking warfarin. (Proteins C&S levels drop to 40-60% within 48 hours of Warfarin initiation and increase to 70% after 2 weeks) – In pregnancy, both free and bound Protein S is decreased. – Also seen in liver disease. |
|
|
Protteiin S - testing issues, treatment
|
Protteiin S
• Testing difficult - technical problems, interference (Heparin, Warfarin, clotting) • Treatment – Heparin for acute thrombosis. – Maintenance anticoagulation with warfarin or low dose subcutaneous heparin. – Prophylactic heparin during pregnancy and delivery. – Peri-operative prophylaxis with heparin. |
|
|
Fibrin Degradation Productts (FDP’s)
|
Fibrin Degradation Productts (FDP’s)
• Activation of Fibrinolytic pathway occurs with deposition of fibrin, as counterbalance to coagulation cascade • Deposited clot incorporates inactive Pllasmiinogen, bound to lysine residues from fibrinogen conversion to fibrin. • Tiissue Pllasmiinogen Acttiivattor (TPA) released from endothelial cells, and converts plasminogen to pllasmiin • Plasmin degrades fibrin into soluble fragments (FDP’s) which are released into circulation. Plasmin also digests other clotting factors (eg Factor V, VIII) and interferes with platelet function • Elevated FDP’s indicate activation of coagulation and fibrinolytic pathways eg thromboembolism, DIC |
|
|
Circulating Anttiicoagullantts
|
Circulating Anttiicoagullantts
• Endogenous substances that inhibit coagulation • Usually antibodies that neutralise clotting factors (eg antibodies versus Factors VIII or V) or activity of procoagulant phospholipids in coagulation tests (eg Lupus anticoagulant) • Rarely may be glycosaminoglycans with Heparin like actions due to increased AT III activity eg Multiple myeloma, haematological malignancies) |
|
|
Lupus Anttiicoagullantt and Anttiicardiiolliipiin
Anttiibodiies - misnomer?, causes? |
Lupus Anttiicoagullantt and Anttiicardiiolliipiin
Anttiibodiies • Really a misnomer, as patients’ with lupus anticoagulant or anticardiolipin antibodies present are at significantly increased risk of thromboembolic disease for reasons that remain unclear. – eg. Antiphospholipid syndrome • These substances react with phospholipids used in coagulation tests to prolong the results eg aPTT suggesting an anticoagulant state • Lupus anticoagulant associated with – Systemic Lupus Erythmatosus (SLE) , – other autoimmune diseases, – drugs eg • antibiotics, • cocaine, • hydralazine, • phenothiazines, • phenytoin, • procainamide, • quinine, • quinidine |
|
|
Anttiicardiiolliipiin Anttiibodiies
Anticardiolipin antibodies • Clinical Features |
Anttiicardiiolliipiin Anttiibodiies
Anticardiolipin antibodies • Clinical Features – Thrombosis and Thromboembolus • Recurrent DVT (intracranial veins, retinal veins, upper extremities, hepatic veins, portal veins, renal veins, inferior vena cava, lower extremities) • Pulmonary embolism • Arterial thrombosis (coronary arteries, retinal arteries, brachial arteries, mesenteric arteries, peripheral arteries, aorta). – First thrombotic often occurs in early-mid teenage years. – Women may experience recurrent foetal loss, usually in second or third trimester. – Migraine headaches, transient ischemic attacks, or Guillain-Barre syndrome is often seen. – Mild thrombocytopenia is often seen. – There is often a dermatologic condition known as livido reticularis in which the skin appears mottled. This is usually most notable in the lower extremities. – There is often a postpartum syndrome, which occurs 2-10 weeks after delivery and consists of fevers, dyspnea, pleuritic chest pain, pulmonary infiltrates, pleural effusions, cardiomyopathy, and arrhythmias. |
|
|
Anticardiolipin antibodies - possible mech of disease, lab measurements, treatment
|
Anttiicardiiolliipiin Anttiibodiies
Anticardiolipin antibodies • Possible mechanisms for anticardiolipin mediated disease – Interaction with platelets to activate their membrane phospholipids, thus initiating the coagulation cascade. – Interference with endothelial release of prostacyclin. – Interference with activation of Protein C on thrombomodulin. – Interference with Antithrombin III activity. – Interference with endothelial release of plasminogen activator. Laboratory Measurements- Anticardiolipin Antibodies • IgG, IgA, and IgM anti-cardiolipin antibodies or anti-phospholipid antibodies can be measured. • The presence of any one antibody (G, A or M) is a risk factor for thrombosis Treatment • short term anticoagulation with heparin and long term anticoagulation with warfarin |
|
|
Lupus Anttiicoagullantt
Clinical features - labtesting, treatment |
Lupus Anttiicoagullantt
Clinical features Lupus Anticoagulant - similar to anticardiolipin antibodies – Thrombosis and thromboembolism (as above) – False positive VDRL – Thrombocytopenia – Recurrent foetal loss – Prothrombin deficiency – Livido reticularis Laboratory Testing Two tests are used: • aPTT with 1:1 mixture of patient's plasma and normal pooled plasma If the aPTT continues to be prolonged even after mixing, there is some sort of inhibitor, either against a coagulation factor or against phospholipids. • Modified Russell Viper Venom Time (MRVVT)- snake venom can activate Factor X, bypassing the need for all coagulation factors except factors V, X, prothrombin, and fibrinogen. Dilute phospholipid is used to increase the sensitivity in looking for lupus anticoagulants. Treatment • short term anticoagulation with heparin and long term anticoagulation with warfarin |
|
|
Disseminating Intravascular Coagullattiion
(DIC) - physiology - causes |
Disseminating Intravascular Coagullattiion
(DIC) Physiology • syndrome that occurs when the clotting cascade goes awry. • clotting and bleeding disorder that results from the generation of tissue factor activity within the blood. • This trigger of the coagulation cascade quickly leads to significant thrombin production which perpetuates its own formation. • In very little time, the existing regulatory factors such as antithrombin III, protein C, and protein S are consumed. • large amounts of thrombin are generated, leading to a hypercoagulable state. • the quantity of plasmin is significantly increased, leading to the generation of significant quantities of fibrin degradation products. This often results in bleeding. Causes • Complications of obstetrics where uterine material with tissue factor activity gains access to the maternal circulation such as in abruptio placentae. • Infection with gram negative bacteria which secrete an endotoxin that induces the generation of tissue factor. • Malignancy - particularly adenocarcinoma of the pancreas or prostate as well as promyelocytic leukemia. • Another less common cause is head trauma. |
|
|
Disseminating Intravascular Coagullattiion
(DIC) Clinical features sub acute and acute |
Disseminating Intravascular Coagullattiion
(DIC) Clinical features • Subacute DIC – associated with thromboembolic complications such as DVT and PE as well as with vegetations on heart valves. • Acute DIC – Thrombocytopenia and depletion of coagulation factors leads to bleeding tendency. – worsened by increased degradation of fibrin to fibrin split products which interfere with fibrin polymeration and with platelet function. – Fibrin deposition into small blood vessels leads to tissue ischemia. The most vulnerable organ is the kidney, where fibrin deposition can lead to acute renal failure. – Hemolysis can occur through mechanical damage to the red blood cells secondary to the fibrin deposits. – Patient may experience neurological phenomena caused by ischemic injury to the brain. |
|
|
Disseminating Intravascular Coagullattiion
(DIC) Laboratory findings acute and subacute |
Disseminating Intravascular Coagullattiion
(DIC) Laboratory findings • Subacute DIC – Thrombocytopenia – Normal or slightly prolonged aPT – Short aPTT – Decreased fibrinogen – Increased fibrin split products • Acute DIC – Thrombocytopenia – Markedly prolonged aPT and aPTT (over 200 seconds) secondary to insufficient fibrinogen. – Very high levels of D-dimers and fibrin split products – Low levels of the specific clotting factors |
|
|
Disseminating Intravascular Coagullattiion
(DIC) Treatment of DIC |
Disseminating Intravascular Coagullattiion
(DIC) Treatment of DIC – Identify and treat the underlying cause – Uterine evacuation for abruptio placentae – Broad spectrum antibiotics for gram negative sepsis – Replacement therapy with cryoprecipitate (for coagulation Factors and fibrinogen) and platelets – Uses of heparin • Can be used if thrombotic complication (eg. oliguria secondary to acute renal failure or cyanosis of digits) • Can use heparin to prevent thrombotic complications if DIC is occurring secondary to a malignancy that is not quickly treatable. • Avoid in patients with head injury or with CNS bleeding. |
|
|
Effects of anticoagulants on tests for hypercoagulability
|
Effects of anticoagulants on tests for hypercoagulability
Test Can be performed whilst patient on Heparin Can be performed whilst patient on warfarin Antithrombin III* Yes Yes Protein C Yes No Protein S Yes No Anticardiolipi n antibodies Yes Yes Factor V PCR Yes Yes DNA test & Prothombin Gene Yes Yes • Heparin may lower antithrombin III levels. If levels are normal whilst patient is receiving heparin the test need not be repeated. Low levels, whilst patient is on heparin, should be repeated. |
|
|
Diiagnosiis of Thromboembolliic Diisease
clincial signs and symptoms investigations |
Diiagnosiis of Thromboembolliic Diisease
• Clinical signs and symptoms – swelling of limb, redness and inflamation for DVT – lung signs for PE (eg SOB, Haemoptysis, pleuritic pain) – ECG changes in severe PE cases • Investigations – Venogram for DVT - Ultrasonic examination; flow studies – V/Q Scan for PE – CT Pulmonary Angiogram – Lab evaluation - coags, D-Dimer SR, etc |
|
|
CT Pulmonary Angiogram
|
CT Pulmonary Angiogram
CT PA • is a medical diagnostic test that employs computer tomography to obtain an image of the pulmonary arteries. • introduced in the 1990s as an alternative to VQ scanning, which relies on radionuclide imaging of the blood vessels of the lung. • It is regarded as a highly sensitive and specific test for pulmonary embolism. Method • 50-150 ml of radiocontrast is injected by syringe driver at a rate of 4 ml/second. • Generally the scan commences automatically when the contrast is detected at the level of the proximal pulmonary arteries or about 10-12 seconds after the injection has started. • Slices of 1-3 mm are performed are 1-3 mm intervals, depending on the nature of the scanner (single- versus multidetector) |
|
|
CT PA
Interpretation and advantages |
CT PA
Interpretation • On CTPA, the pulmonary vessels are filled with contrast, and appear white. Any mass filling defects (embolus or other matter such as fat or amniotic fluid) appears darker. • Generally, the scan should be complete before the contrast reaches the left side of the heart and the aorta, which could result in artifacts • an embolus classically produces a filling defect within the affected pulmonary artery. Non-occlusive emboli have a "tram-track" appearance. • Although considered the gold standard, angiography may not always detect the presence of emboli. Some indirect angiographic evidence for the presence of emboli such as vascular pruning and delayed capillary blush are non-specific. Advantages (cf V/Q Scan) • rapid test • Widely available • No radioactive nucleotides used • Equal results to V/Q scan |
|
|
Plasma D-Dimer.
|
Plasma D-Dimer.
• Activation of the Coagulation cascade also activates the Fibrinolytic pathway as a counterbalancing mechanism to prevent excessive intravascular thrombosis. • Fibrinolytic pathway activation leads to increased production of the protein D-Dimer • D-Dimer is a degradation product of cross-linked fibrin generated by plasmin cleavage. The plasma D-dimer level is a sensitive indicator of fibrinolysis. • In the context of activation of the coagulation cascade, Normal levels of the protein have high negative predictive value; that is, a patient whose D-dimer level is less than 400 Xg per litre is unlikely to have embolic disease. • Increased D-dimer level does not have a strongly positive predictor value as anything which activates the fibrinolytic pathway will elevate the level. • eg infection, inflammatory diseases, necrosis, cancer, trauma • Essential to evaluate D-Dimer value in combination with clinical symptoms to make it a useful tool. |
|
|
D-Dimer Simply Red
|
D-Dimer Simply Red
• D-Dimer SR is a commercial reagent that verifies elevations in D-Dimer • Positive D-Dimer SR is a useful rapid qualitative test for embolic disease (compared to quantitative test), and may indicate need perform other tests eg V/Q scan, CT PA, venogram • Needs to be considered in combination with clinical symptoms |