![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
236 Cards in this Set
- Front
- Back
|
What are the types of post-translational modications in the ER? |
1. Glycosylation (adding carbs) 2. Protein folding (Ex. BiP) 3. Disulphide bond formation (Ex. PDI) 4. Proteolytic cleavage |
|
|
Where are the two types of possible proteins modified? |
1. Proteins targeted to the ER lumen -Modified along the entire length of the protein 2. Proteins targeted to the ER membrane -Modified on the lumen portion of the protein -Not modified on the cytosolic portion or transmembrane domain |
|
|
Many types of glycosylation but most common is N-linked. What is N-linked glycosylation? |
-Process of adding a sugar/polysaccharide to NH2 of the R-group of asparagine |
|
|
How does the process work? What is the outcome? |
Enzyme OSTase adds polysaccharide to protein --Polysaccaride stays on LUMINAL side of the ER also known as the ectoplasmic side although embedded in the membrane |
|
|
Protein folding; Lectin example & BiP (Hsp40) |
-comprised of calreticulin & calnexin -Can recognize modified proteins and fold them as do molecular chaperones |
|
|
Explain disulphide bond formation in cytoplasm and ER |
Cytoplasm --Reducing environment --FAVOURS taking away disulphide bonds
ER--oxidizing environment --FAVOURS creating disulphide bonds |
|
|
Are proteins that will be secreted from ER or stay in ER more likely to have disulphide bonds? |
Secreted |
|
|
What is an example of a molecule that needs its disulphide bonds? |
Pancreatic ribonuclease A (RNAase A) -Four disulphide bridges -Maintains functional state of the enzyme even in acidic conditions |
|
|
How are disulphide bonds formed? |
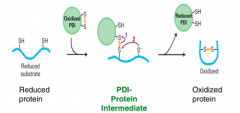
1. Two cystine residues
2. Oxidized PDI w/ disulphide bridge reacts w/ one cysteine 3. Oxidized protein is formed and reduced PDI is in environment |
|
|
Proteolytic cleavage |
-N-terminal signal cleavage -Cleavage of the peptide backbone of a protein -All TYPE I integral membrane proteins have the N-terminal sequence cleaved by a signal peptidase which is required for folding |
|
|
How to solve the problem when there are too many unfolded proteins in the lumen?
UPR!!!! |
STEP 1 -Slow translation of new protein translation or removing unfolded proteins of the ER for degradation
STEP 2 (UPR target genes) -Increase production of proteins needed for protein folding |
|
|
How does step 1 of UPR work? |
-BiP dissociates from Ire1 when unfolded proteins are present b/c it has a high affinity for them -BiP acts as chaperone and assists in folding while Ire1 makes homeodimers which act as endonucleases (which will clip the polypeptide backbone req'd for folding) -Ire1 dimers cut HAC1 which is what is acting to inhibit translation -Hac1 transcription factor is sent to nucleus where it will activate genes like BiP, PDI, lectin and signal peptidases |
|
|
Anterograde and retrograde transport |
-From ER toward cell membrane in vesicles |
|
|
Three techniques for how a protein gets from the ER out of the cell? |
1. Pulse-chase labelling 2. Fluorescent microscopy of GFP-labelled proteins 3. Genetic mutations that disrupt transport |
|
|
Explain pulse-chase and system in mammalian cells |
Pulse: Brief incubation using methionine in medium w/ radioactive amino acids --Brief so NOT all of them get labelled
Chase: Unlabeled medium --Washed and transferred to non-radioactive medium so you can see the labelled things
System: Acinar cells of pancreas |
|
|
Explain the different locations among different times |
PULSE ALL SAME TIME (3 minutes)
CHASE 0: ER, 17: Golgi complex 117: Secretory vesicle |
|
|
Tracking GFP-tagged secreted proteins |
-Researchers targeted and tracked a viral protein that is encoded in the viral genome but synthesized in the host cell membrane. Protein is apart of the viral envelope that surrounds the virus if needed for infection |
|
|
Pulse, chase and system of tracking GFP-tagged secreted proteins |
Pulse -Brief incubation in medium w/ radio active amino acids
Chase -Unlabeled medium
System -VSV g-glycoprotein |
|
|
Explain the temp change |
32: permissive temperature; protein folded and transported to ER membrane
40: restrictive temperature; misfolded protein, retained in ER --Protein variant denatures and is retained |
|
|
Explain the pulse chase results w/ times |
0: ER, 40: Golgi, 180: Plasma membrane |
|
|
Explain screen and system in genetic analysis of secretory pathway |
1. Screen: Identify genes req'd for protein secretion in yeast
2. System: Synthesis and transport of the secreted protein invertase in yeast |
|
|
Two steps of secretary pathway in yeast cells |
1. Converts sucrose to
Glucose and fructose by invertase 2. Both glu and fruc imported into the cell |
|
|
What happens when there is a defect? |
Failed secretion of invertase and accumulation of vesicles (invertase is reaction that converts glucose to fructose) |
|
|
Temperature sensitive mutations in genes of secretory mutants |
Random mutations and soundness that at permissive temp, protein would fold but didn't at the restrictive temp --At restrictive temperature there is a buildup of invertase in secretory vesicles |
|
|
Explain what clumps at diff steps |
Invertase
|
|
|
What can cause Class A sec mutants |
SRP, SRP receptor, signal sequence of invertase, sec61alpha-ER translocon |
|
|
CLASS A MUTANTS |
Fate of secreted protein: -Accumulation in the cytosol
Defective function: -Transport into the ER |
|
|
CLASS B MUTANTS |
Fate of secreted protein: -Accumulation in the rough ER
Defective function: -Budding of vesicles from the rough ER |
|
|
CLASS C MUTANTS |
Fate of secreted protein: --Accumulation in the ER-to-Golgi transport vesicles Defective function: -Fusion of transport vesicles w/ golgi |
|
|
CLASS D MUTANTS |
Fate of secreted protein: -Accumulation in golgi
Defective function: -Golgi to secretory vesicles |
|
|
CLASS E MUTANTS |
Fate of secreted protein: -Accumulation in the secretory vesicles
Defective function: -Transport from secretory vesicles to cell surface |
|
|
TWO pathways |
1. Constitutive Secretory Pathway -Vesicles fuse with the cell membrane to release proteins from the process called exocytosis -Used by proteins that are released immediately after protein synthesis and transport
2. Regulatory secretory pathway -Kept in cell until a signal triggers release
3. Lysosome |
|
|
What is used to highlight the golgi complex |
Wheat germ agglutinin -used to recognize the Golgi complex -A lectin that recognizes N-linked polysaccharides found in Golgi cisternae |
|
|
Components of the golgi complex |
trans-golgi network, trans cisternae, medial cisternae, cis cisternae, cis-golgi network |
|
|
Golgi cisternae |
(trans cisternae, medial cisternae, cis cisternae)
themselves contain resident proteins are that necessary for further PTM for transported proteins |
|
|
TWO MODELS OF anterograde transport |
MODEL A--Vesicular transport model-- -Cis to med to trans-cisternae
MODEL B--Cisternal maturation mode-- -Proteins stay in cisternae themselves but the actual cisternae are moving forward through the golgi complex -Requires vesicles moving in the retrograde direction |
|
|
Explain the experiment about the models and which one it conclused |
1. Antibody to cell membrane protein that is undergoing anterograde transport --Protein is restricted to golgi cisternae and not found in vesicles according to TEM which means that they stay in cistern 2. Antibody that should only be found in the medial golgi --Resident medial golgi protein that is moving in both directions and is found in vesicles |
|
|
What are the implications of this? |
-New cis-golgi cistern are formed by vesicles from ER -Trans-golgi cisternae dissipate into transport vesicles -Golgi-resident proteins must be resorted in the anterograde direction because they are being used up,this can be done by vesicles in the retrograde direction |
|
|
Explain transport vesicle formation and function |
1. Budding forms the beginning formation of new vesicles from the DONOR compartment 2. Vesicles are loaded with cargo using receptors 3. New vesicle is fully formed and released 4. Vesicle docking and fusion the the membrane of the recipient compartment |
|
|
Coat proteins |
-Control vesicle assembly
|
|
|
What binds to these coat proteins? |
-GTP b/c they are g-proteins with GTPase activity
|
|
|
How to get from active to inactive or vice versa |
Inactive--> Active (GTP in, GDP out needed GEF to do this)
GAP needed to take out GTP |
|
|
Three types of vesicles and what they do |
1. Clathrin vesicles -away from trans-golgi network to endosomes and secretory vesicles (also used in endocytosis) 2. COP I vesicles -Retrograde between golgi and ER 3. COP II vesicles -Anterograde -RER to golgi |
|
|
What is the first step in vesicle formation? |
Sar1 which is inactive will bind to transmembrane protein sec 12, then GEF is used to add GTP which produces a conformation change in Sar1 and activating it as it becomes anchored to the membrane |
|
|
Different types of COPII coat proteins |
Sec 23, 24, 13 and 31 which can all associate with Sar-1 GTP |
|
|
How can they bind to so many COP II coat proteins? |
Sec-23 binds directly while Sec-24 (attached to a cargo receptor which leads to the inside of vesicle) binds indirectly and more proteins accumulate they made the membrane curved |
|
|
ARF |
-Same function as sar-1 but is used in COPI and clathrin-coated vesicles |
|
|
Second step in vesicle formation: How is cargo loaded? |
-Vesicle coat proteins recognize target cargo and cargo receptors and they accumulate inside while some ER resident may sneak it |
|
|
How is COPII vesicle released? |
Sar-1 GTP hydrolysis |
|
|
How is coat disassembled? |
-Sar-1 is no longer anchored to the membrane and is released w/ coat proteins and the uncoated vesicle is carried to the recipient membrane by motor proteins |
|
|
How can you inhibit coat disassembly? How do you know it is being inhibited? |
Accumulation of COPI vesicles; promoted by non-hydrolyzable GTP or a mutation in G-protein |
|
|
Triscallion |
-When three light chains and three heavy chains interact in clatharin vesicles |
|
|
Polyhedral lattice |
Clathrin coat -Clathrin heavy chains -Clathrin light chains -Adaptor proteins |
|
|
Dynamin |
-G-protein req'd for vesicle release in clathrin coated vesicles -Has an Ap complex and fibrous clathrin coat -Released when GTP is hydrolyzed |
|
|
Two models for dynamin function |
1. Pinchase -Suggests that dynamin helices Constrict and squeeze the membrane to initiate release 2. Poppase -Suggests that dynamin helices Elongate and push the vesicle away From the donor membrane |
|
|
Evidence for dynamic function pinchase |
-Dynamin-lipid tubules contract upon GTP addition -Allows for conformational shift and narrows internal diameter -Consistent with pinchase |
|
|
Evidence for dynamic function poppase |
1. Undecorated lipid tubes (no dynamin) 2. Dynamin GTPgammaS on lipid tubes; arressted by no hydrolysis 3. Dynamin GDP on lipid tubes * Increased space between Spirals shows that it is consistent With POPPASE |
|
|
Shibre |
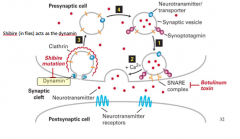
-Acts in flies as dynamin |
|
|
Shibre mutation |
-Is a temperature sensitive mutant in fruit flies and it disrupts the formation of synaptic vesicles and no vesicle release can occur b/c temperature sensitive mutation has been added to dynamin protein
Permissive temp is 20 but restrictive temp is paralyzed at 30 (but reversible) |
|
|
Rab GTPase |
-Rab-GDP found free in the cytosol -Rab-GTP anchored to vesicle membrane and blinded through hydrophobic anchor
|
|
|
Rab effector |
-Anchored to target membrane and recognizes Rab
|
|
|
Are both Rab GTPase and Rab effector needed for docking? |
Yes |
|
|
What is needed for vesicle fusion? |
Vesicle membrane: v-SNARE (Ex. VAMP) Target membrane: t-SNARE (ex. syntaxin, SNAP-25)
These interactions between SNARE result in fusion --Strong complex that allows for fusion |
|
|
How does the membrane fusion work? |
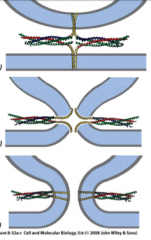
-Vesicle docking (Snare complex is formed) -Speculative transition state (as they spiral they bring them closer) -Membrane fusion--resealing occurs |
|
|
How is SNARE complex disassembled? |
Cytosolic SNARE binding proteins (NSF and a-SNAP) required to dissociate SNARE complexes by unwinding the 4 helices, need ATP to be added and then the membrane can be recycled and reused |
|
|
Why is there retrograde transport? |
1. Recycle membrane v-SNARES 2. COPII vesicle cargo receptor 3. Unfolded proteins 4. Return incorrectly sorted ER-resident proteins |
|
|
Diff signals (1) on soluble ER resident proteins |
Lys-Asp-Glu-Leu (KDEL) |
|
|
Diff signals (2) resident ER membrane proteins |
LysLysXX (KKXX) |
|
|
Diff signals (3) cargo acceptor from COP II vesicle -Needed to load cargo in retrograde |
Asp-X-Glu |
|
|
What does KDEL receptor do? |
-It recognizes KDEL sequence on ER resident protein and loads it into COPI vesicle |
|
|
Explain the mechanism behind unicellular growth |
-Spore germinates and then starves -Unicellular amoebas aggregate due to production of cAMP -Loose and tight aggregates form and slug cells are able to differentiate when nutrition is good -Slug cells form into a fruiting body made out of spores and stalk |
|
|
When does germination happen at unicellular growth occur? |
-When food is abundant |
|
|
What forms the spore and the stalk of the fruiting body? |
-Spore -- posterior end of slug turns into spore
-Stalk --anterior end of slug turns in stalk |
|
|
How do cells aggregate? What is the signal and what is the receptor? |
Signal: cAMP receptor; GPCR g-protein coupled receptor |
|
|
Mutation causing no net movement |
-Clathrin heavy chain -Means that the cells are unable to form the vesicles necessary for transport of proteins To the cell membrane -MOVING BUT NO NET MOVEMENT WHY? -In the absence of protein transport, the transmembrane G-protein coupled receptor is not transported to the cell surface AKA NO RECEPTOR FOR CAMP THE CELL CANNOT RESPOND TO SIGNAL |
|
|
What do human neutrophil cells move towards? |
White blood cells move toward bacteria |
|
|
Signal and receptor of human neutrophil |
Signal: fMLP (unintentionally produced by bacteria Met-Leu-Phe) Receptor: fMLP receptor; g-coupled receptor |
|
|
Cell-cell signaling |
Transmitting information from one cell to another and inducing a change in behaviour of response |
|
|
How does the ligand bind to the receptor? |
Molecular complementarity --Conformational change in the intracellular domain of the receptor which allows for the signal transduction and change in behaviour to occur |
|
|
How is specificity ranked? |
1. Molecular complementarity from
Ligand and binding receptor
2. Specificity of intracellular response That is mediated by Signal transduction pathway |
|
|
Can one signal have different responses? |
-Yes, in different cells have different internal signal transduction pathways |
|
|
Cellular responses (2) |
1. Fast response -Change in enzyme activation through some sort of phosphorylation 2. Slow response -Change in gene transcription |
|
|
Kd |
Concentration where half of the receptors are occupied |
|
|
What is the concentration the produces 50% of physiological response? |
Much lower than concentration L from Kd |
|
|
Why is it lower concentration? |
Because cell signals inside the cell become amplified |
|
|
Secreted signals (2) |
1. Endocrine signaling -SIGNAL: Hormone -TARGET: Very distal 2. Paracrine signaling -SIGNAL: Growth factors, neurotransmitters -TARGET: proximal --through diffusion aka synapse |
|
|
Signaling that requires cell contact (2) |
1. Integral membrane proteins -Come into contact with each other, could be transmembrane proteins on different cells SIGNAL: Membrane bound TARGET: Neighbour 2. Plasmodesmata --Gap junctions in animals, connecting cytoplasms of neighbouring cells SIGNAL: Cytosolic TARGET: Neighbour, but signal can move through cells |
|
|
What are secondary messengers? |
A cell may respond to a primary extracellular signal by producing an internal secondary messenger which can then diffuse from one cell to another to exert the same response and coordinate the behaviour of a series of cells |
|
|
Autocrine signaling |
-Signals with itself
Signal molecule: secreted, ex. growth factors (induce cell division, stop cell division)
Target: receptors on that same cell for that signal |
|
|
Types of effector proteins |
1. Tyrosine kinase-linked receptors/ CYTOKINE RECEPTOR ---Phosphorylation of effector protein
2. Receptor tyrosine kinases ---Phosphorylation cascade
3. G-protein coupled receptors ---Effector enzyme produces second messenger |
|
|
Signal and response in JAK-STAT pathway |
Signal: Erthropoietin (Epo) Response: RBCs Cells proliferate |
|
|
What happens without Epo? |
Erythropoietin progenitor cells undergo apoptosis which leads to cell death |
|
|
Epo receptor |
--Dimerization in presence of Epo, inactive as monomer |
|
|
What are the intracellular transduction pathways that happen due to expo binding to epo receptors? What is the cellular response? |
-JAK kinases and STAT transcription factors -Transcription of STAT target genes happens and apoptosis is then inhibited |
|
|
Three functional domains of EpoR |
1. Cytosolic domain
2. Transmembrane domain 3. Extracellular domain |
|
|
What is the process of receptor dimerization? |
-Signal binding; ligand binds to binding sites -Dimerization and phosphorylation of activation lip tyrosine which activates JAK kinase -Phosphorylation of docking sites |
|
|
What is STAT transcription factor made up on? |
SH2 domain and DNA binding domain |
|
|
What happens after STAT transcription factor binds to phosphorylated sites? |
-STAT with phosphate group binds with another and is sent to nucleus where it will bind to DNA and activate transcription factors |
|
|
P-Tyr sequence on SH2 protein binding |
Pro-Asn-pTyr-Glu-Glu-IIe-Pro |
|
|
SH2 domains recognize...
PDZ domains recongize... |
-Phosphotyrosine
-hydrophobic C-terminus |
|
|
What else binds to phosphorylated tyrosine |
SH2, PTB and 14-3-3 --good b/c reversible |
|
|
What doesn't binds to phosphorylated tyrosine |
PDZ, SH3, WW |
|
|
What happens after STAT is activated |
Increases transcription of Bcl-Xl which prevents apoptosis |
|
|
Why is it good that this pathway can be turned off? and how? |
-Increased viscosity of RBCs can make blockage or narrow capillaries
1. Reversing phosphorylation ----SHP1 (SHORT TERM) 2. SOCS protein
(LONG TERM) |
|
|
SHP1 Phosphatase |
-Has two SH2 domains and it binds to phosphate on JAK and at docking sites preventing the transcription factor STAT from binding |
|
|
Protein degradation by SOCs, when does this happen? |
--Used when oxygen levels are high in the body --Binds to the phosphorylated Docking sites via SH2 domain --E3 ubiquitin ligase is recruited and it targets JAK kinase for degradation and turns off the signalling pathway |
|
|
Mutations in EpoR gene |
-Truncated versions of the Epo receptor (proteins that have shorter docking sites) ----Not as much is phosphorylated, more apoptosis
-Decreased sensitivity to SHP-1 and SOCS --Already apoptosis so no dramatic effects
-Similar to Epo doping RBC counts |
|
|
What does RTK bind to? |
Nerve growth factor (NGF), Platelet derived growth factor (PDGF), Epidermal growth (EGF), Insulin |
|
|
What responses does RTK have? |
-Cell differentiation, division, apoptosis, metabolism |
|
|
What is the pathway of RTK receptor? |
Ligand growth hormone binds to RTK which the adaptor proteins (GRB2) and Ras (connected) bind to phosphate groups --In order to activate Ras, effectors are needed which are GEF and GAP --MAP kinase is activated when it is phopsphorylated --Dimerization through ATP and autophorphorulation occurs and cellular response happens |
|
|
How is RTK activation? |
-Ligand binds -Dimeriztion and phosphorylation of activation lip tyrosine (Get brought together) and phosphorylated through ATP -Phosphorylation of additional tyrosine residues because of autophosphorylation |
|
|
Adaptor proteins |
-Carry two or more protein interaction domains that allow the proteins to act as linkers between other proteins -Indirectly linking proteins to the receptor |
|
|
Adaptor protein examples |
1. IRS-1; PTB domain binds to the phosphorylated tyrosines and as well as the SH2 domain |
|
|
Scaffold proteins |
--Multiple protein-protein interaction domains and assemble proteins in an ordered series |
|
|
Explain the organization of the adaptor protein |
-Three protein-protein domains
1. One SH2 domain and two SH3 domains
SH2 -recognizes phosphoryalted tyrosines and binds to RTK
SH3 -Recognizes pro-rich sequences and binds SOS |
|
|
How is RAS protein inactivated? |
-Inactivated by GDI ----Increases the affinity of the nucleotide-binding pocket for GDP keeping the G-protein inactive OFF |
|
|
What does SOS bring to membrane? |
Ras inactive and then because GTP is naturally high concentration it replaces and activates RAS |
|
|
What does Ras-GTP active do? |
-Results in cell division
|
|
|
NF1, loss of function |
-enhances the intrinsic GTase activity of RAS, accelerating the rate of hydroloysis of GTP, inactivating RAS (shortening time that is active)
Loss of function means that GTP cannot be hydrolyzed as fast and then cell division keeps occurring |
|
|
Four hypotheses if Ras and RTK are needed for cell diviion |
FOUR HYPOTHESES 1. Ras upstream of RTK2. Ras downstream of RTK 3. Both not needed 4. Both needed for cell division |
|
|
Is Ras downstream of RTK? experiment |
1. EGF (ligand) + EGF receptor = proliferation 2. EGF + Ras antibody (blocks Ras) = no proliferation 3. No EGF, RasD mutation fails to hydrolyze GTP = tumorogenesis |
|
|
Conculsion of Is Ras downstream of RTK? experiment |
Yes, downstream ras activation happens after rtk activation |
|
|
How can you disrupt the RTK pathway? |
-Failure in GTPase activity of Ras -Failure in GAP protein (NF1) -Always active receptor tyrosine kinase linked to breast cancer (Her2) (never responds to EGF signal)
|
|
|
How does Ras activation lead to Raf activation? |
1. Ras is phosphorylated 2. Inactivate raf (due to 14-3-3 adaptor protein which is bound to Raf) attacks Ras and releases 14-3-3 to activate raf 3. Hydrolysis leads to dissociation from Ras from Raf |
|
|
What does Raf do once it is activated? |
-Leads to activate MEK -Phosphorylates MEK at MEK's tyrosine and threonine kinase sites |
|
|
How does MEK phosphorylate MAPK? |
-At Serine/theronine kinase sites - --Dimerizes upon activation and is translocated to the Nucleus where target transcription factors are Phosphorylated and activated |
|
|
How does this happen in order? |
--Scaffold proteins to hold together |
|
|
Where is MAP kinase kinase phosphorylated? |
-Specific The (T183) and Tyr (Y185) -Induces a conformational change to activate the |
|
|
How is transription activated? |
-Inactive p90RSK is phosphorylated by dimeric MAP kinase dimer (ERK1 AND ERK2) -Phosphorylation of two transcription factors 1. TCF directly by MAP kinase when they are both inside the nucleus 2. SRF directly phosphorylated by p90 RSK inside the nucleus
-Trimer (TCF + 2SRFs bind to the SRE element on several genes) including c-fos
|
|
|
What is SRE element? |
-Enhancer sequence which promotes the assembly of RNA polymerase and transcription of the target gene |
|
|
G-protein coupled receptors |
STRUCTURE: -7 transmembrane alpha helices that loop through the membrane of the cell (span the membrane 7 times) -4 extracellular segments E1 through E4 which will fold in the extracellular space to form the signal Binding domain -4 cytoplasmic segments C1 through C4 which will fold to form an internal domain that interacts with a trimeric G protein |
|
|
Explain adrenergic stress response in mammalian cells |
Signal: catecholamines Receptor: GPCR Intracellcular transduction: Adenyl cyclase and cAMP Cellular response: release of stored energy req'd for fight or flight response |
|
|
What are catecholamines? |
Epinephrine (Adrenaline), norepinephrine (noradrenaline) and dopamine |
|
|
What are the two responses that epinephrine responds to? |
1. Beta-adrenergic receptors: STIMULATORY 2. Alpha-adrengergic receptors: INHIBITORY |
|
|
Beta-adrenergic receptors: STIMULATORY |
-Liver and adipose cells: glycolysis and lipolysis -Heart muscle: Increase contraction, increase blood supply to tissues -Smooth muscles cells of intestine: increase relaxation, save energy for major locomotary muscles |
|
|
Alpha-adrengergic receptors: INHIBITORY |
-Blood vessels of smooth muscle of intestine, skin kidney: causes arteries to constrict, blood supply is reduced to periphery |
|
|
Why is it good that one signal has multiple effects? |
-Can coordinate multiple responses |
|
|
How is receptor activated in stress response? |
1. Binding hormone induces a conformation response and opens up a hole for the G subunit to bind 2. Activated receptor binds to G-beta-gamma of G-subunit 3. G-beta-gamma dissocaites and is replaced by GTP allowing G-protein to move laterally and active while g-protein is associated |
|
|
What is the effector enzyme? |
Adrenylyl cyclase |
|
|
Explain concentrations of cAMP and what effects it within beta adrenergic receptors? |
-Gs coupled protein (subunits consist of a, B, gamma) -Gs,alpha-GTP dissociates from Gbgamma and then binds and activates adrenylyl cyclase -Gs, alpha, GDP reassociates with G, beta, gamma and thus inactivating adenylyl cyclase which DECREASES the concentration of cAMP |
|
|
Explain concentrations of cAMP and what effects it within alpha2 adrenergic receptors? |
-Same Gbeta and G-Gamma subunits -Different Galpha, inhibitory Galpha which inhibits adenylyl cyclase (b/c interacts w/ a different region) |
|
|
Why does the concentration of cAMP actually decrease instead of staying the same in beta? |
B/c it is degraded by phosphodiesterase to 5'AMP |
|
|
What does cAMP do? |
Binds to PKA to make it active |
|
|
What happens when cAMP binds to PKA? |
Binds and removes catalytic sites which make it active |
|
|
What does PKA target? |
Glycogen: major storage form of glucose (needed to release ATP)
|
|
|
How is glycogen synthesized and degraded? |
Synthesized by glycogen synthase Degraded by glycogen phosphorylase |
|
|
Net affect of glycogen release in muscle |
-Glycogen to glucose-6-phosphate (G-6-P) which can be used in glycolysis and produce pyruvate and NADH for ATP production |
|
|
Liver
--How is glycogen synthesis inactivated? |
--Phosphorylation and inactivation of glycogen synthase through PKA |
|
|
How is glycogen broke down? |
Indirectly activation of glycogen phosphorylase that degrades glycogen |
|
|
What happens with all the G-6-P that is now available because it was broken down from glycogen? |
-Converted to free glucose and released to blood and transported to other tissues |
|
|
How is gene transcription activated? |
-PKA catayltic subunits phosphorylate CREB which are then dimerized and used to bind to CRE |
|
|
What is basal transcription machinery? |
Holds on to dimer and helps with transcription |
|
|
Monomer and size of fibres |
Actin --Monomer: actin filaments
Microtubules --Monomer: a and b tublin
Intermediate filaments --Different protein sets |
|
|
Filament distribution |
-Actin used to form microvillus -Microtubules form networks -Intermedieate filaments span surface for structural support |
|
|
Motor proteins: AF-based and MT-based |
AF-based: myosin MT-based: kinesin and dyenin |
|
|
Actin based structures and movement |
1. Microvillis 2. Cytoplasmic contractile bundles needed in muscles 3. Needed for cell migration (falipodia) 4. Cell division (directs cytokinesis) |
|
|
Monomer of single actin |
G-actin |
|
|
Explain g-actin monomer |
4 DOMAINS and large cleft between 2 and 4 for ATP binding --The ATP binding pocket is pointed to the minus end of the elongating Polymer so that the ATP-binding pocket of each monomer is not exposed except For the pair of monomers at the very end
POINTED minus-end and BARBED plus-end |
|
|
F-actin depolymerization and polyermization |
At plus end (barbed) polymerization rate is greater than depolymerization rate and at minus end depolymerization rate is greater than polymerization --Strand is usually formed of ADP because it has instrinsic ATPase activity which hydrolyzes |
|
|
Critical concentration |
-Polymerization still happening but no net growth |
|
|
What is associated with dynamics of actin polymerization and depolymerization? |
1. Profilin binds to actin-GTP
-This promotes GTP binding and activating the monomer -Profilin-actin dimers accumulate at the plus-end, increasing the local concentration of active actin monomers -Thymosin binds to actin monomers and inhibits polymerization -Thymosin-actin dimers also accumulate at the plus end creating a buffer of stored actin monomers
2. Capping proteins on the ends of actin filaments can inhibit polymerization and depolymerization |
|
|
Treadmilling |
-Rate of polymerization at plus end Is the same as the rate of depolymerisation at the minus end -NO NET INCREASE IN TOTAL LENGTH -STILL MOVEMENT BECAUSE THE RELATIVE POSITION IS CHANGING |
|
|
Myosin motor proteins |
are able to move along actin filaments and power intracellular cargo trafficking |
|
|
4 types of motor proteins |
I: Same motor domain as II and V, shortest tail domain II: Same motor domain as I and V, longest tail domain III: Different motor protein, second longest tail domain V: Same motor domain as I and II, third longest tail domain |
|
|
What is in the tail domain of myosin I, II and V |
-All have a site for N-terminus that binds actin filaments |
|
|
Myosin II |
-Contains two heavy chains that form a coiled-coil motif and four light chains of two distinct types that form the head shape |
|
|
How is myosin assembled? |
-Polymerization done by myosin tails -By MLC kinase -Assembly of 15-20 myosin II proteins -Motor heads are exposed for association with actin filaments |
|
|
Contractile muscles; Sacromere layout |
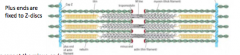
-Thick filaments + actin filaments -Plus ends are fixed to Z-discs
|
|
|
Capping at sacromeres |
-Tropomodulin caps at the minus-ends And CapZ at the plus end |
|
|
Nebulin in sacromeres |
Nebulin binds together parallel actin filaments |
|
|
Where are thick filaments in sacromeres? |
Middle of the sacormere and attached to the Z-discs by a spring protein called titin |
|
|
How do sarcomeres move? |
-The actin filaments are pulled past the myosin filaments toward the middle of the sarcomere by the cyclical association with the myosin motor heads -Shortens without any changes in thickness or thinness -Moves toward plus side of the thin filaments -Causes muscle contraction -Fueled by cylcing through ATP to ADP
-When the myosin thick filament releases the actin thin filaments upon dissociation of calcium from the actin filaments they slide past one another to allow sarcomere elongation and muscle relaxation |
|
|
How does the mechanical cycle work of myosin? |
-Myosin is attached to the actin filament -ATP is added and then myosin-ATP is released -Hydrolysis happens and there is a change in conformation (straight up w/ ADP and Pi) -Pi is removed and then ADP myosin is attached to the actin filament again
-Toward the plus end |
|
|
Myosin V protein |
-Powers intracellular trafficking of cargo along actin filaments -Transport of melanosomes |
|
|
Examples of melanosomes |
-Membrane-encloed organelles containing pigment grannules called melanin -Melanin protects from tanning that may result in DNA damage
EXAMPLE melanocytes (Skin cells) have several dendrites that stretch out to connect it with many keratinocytes |
|
|
What does the rate of myosin protein depend on? |
1. The rate of ATP hydrolysis by ATPase
2. The proportion of time myosin is bound to the Actin filament |
|
|
What does step-size depend on? Give examples |
-Myosin V lever is 3 times longer than myosin II and mysosin V has greater step size -Myosin 5 steps in hand over hand motion: the trailing head detaches from actin and is propelled towards the barbed end during the power stroke of the leading head and the trailing head becomes the new leading head |
|
|
Microtubule filament structure |
-Protofilaments are staggered so that if you follow a string they appear to spiral -Forms ring from birds eye view |
|
|
Explain the heterodimer of microtubules |
-alpha-tubulin is closer to the minus end and doesn't get hydrolyzed although they both have pockets for GTP |
|
|
Explain rescue and catastrophe |
Catastrophe -A-b-GDP are removed -Happens when most of the molecule consists of a-b-gdp dimers
Rescue -A-b-GDP are added to the plus end
|
|
|
EB1-GFP |
EBI-GFP is a plus end binding protein that prevents premature catastrophes and acts as a positive regulator of microtubule growth |
|
|
Dynamic instability
What promotes stability? What promotes instability? |
-Oscillating behaviour of growth and shortening
-Stability is GTP-cap because it gives many a-b-GTP dimers and is maintained at a constant level -Instable would be GDP cap b/c less affinity |
|
|
MAPS and two types |
-Proteins that control the assembly and disassembly of microtubules -Interconnect microtubules to form bundles of cross bridges
1. Stabilization 2. Destabilization |
|
|
Explain MAPS: Stabilization |
EXAMPLE: Tau and EB1 --Frequency of catastrophes suppressed and growth rate is enhanced
RESULT: Longer, less dynamic microtubules |
|
|
Explain MAPS: Destabilization |
EX. Catastrophin --Frequency of catastrophes increased
RESULT: shorter, more dynamic microtubules |
|
|
Gamma-tubulin |
-Involved in the nucleation of microtubules -Less concentration of these -This and gamma-tubulin ring form gamma-TuRC |
|
|
gamma-TuRC |
-Nucleates at the minus end of new microtubules by forming a template for the growing, plus-end -Acts as a cap of the minus end while microtubule growth and dynamics occur at the plus end |
|
|
Where does nucleation happen? |
MTOC --called a centrosome --Formed by two centrioles and a cloud of material that contains multiple gamma-TuRC complexes |
|
|
How does nucleation look in microtubule? |
+ end toward periphery - End is nucleated at the γ-TuRC complexes |
|
|
How does nucleation look in mitosis? |
-As replicated MTOCs separate, microtubules are nucleated at γ-TuRC complexes and the plus-ends grow outwards -Happens at MTOCs |
|
|
Where do the plus ends go? |
-Some of the microtubules plus ends emanate towards the cell periphery and achnor the spindle while others grow towards each other to create the spindle and attach to the condenses replicated chromosomes |
|
|
Microtubule toxins (2) |
1. Colchicine -Inhibits polymerization -Binds and stabilizes free ab-tubulin dimers
2. Taxol -Binds to B-tubulin and increases the affinity Of the dimer for the plus end, preventing depolymerization and stabilizing the microtubules -The effect of this is to prevent the assembly of the mitotic spindle and inhibit mitosis -Used in cancer treatment, ''paclitaxel'' |
|
|
Microtubule motors and which direction to they go in? |
Kinesin family -Move towards the plus end
Dynein family -Move toward the minus end |
|
|
How does the mechanics work behind kinesin? |
Lagging head has ATP while leading head has ADP --Lagging head is hydrolyzed and they are both ADP but then ADP on the leading head is removed and the inorganic phosphate but ATP is added which allows for a conformational change and the lagging head to swing forward |
|
|
Dynein and two types |
Dynein -Minus-end directed motor -Two identical heavy chains and a variety of intermediate and light chains
TWO TYPES 1. Cytoplasmic dynein-Associated w/ microtubules that direct movement of organelles and vesicles In the cytoplasm 2. Axonemal dyenin -Found in structures that the power the movement of whole cells such as Cilia and flagella |
|
|
How does the power stroke work in dynein? |
1. Step 1: ATP binding releases the motor head group from the microtubule 2. Step 2: ATP hydrolysis; dynein- ADP+ Pi can now attach to the microtubule 3. Step 3: Release of Pi powers the power stroke of the linker |
|
|
Movement of cargo is driven by power of what? |
LINKER |
|
|
Explain bidirectional movement of vesicles |
-MTOC as minus ends of the cell body and plus ends toward the periphery and the synapse -Anterograde transport would be toward the synapse |
|
|
How does it end of moving if it's pulling both ways? |
MODEL 1---- Tug of war, final direction of movement is decided by the winner of the battle REGULATORY PROTEINS THAT CONTROL DIRECTION IN RESPONSE TO SINGALS WITHIN THE CELL |
|
|
Melanosomes |
-Movement of the organelles changes the colour of the skin cells in response to behavioural signalling |
|
|
How do dynein and chines relate to skin cells? |
1. DYNEIN
-Concentrate the melanosomes at the center -Move the melanosomes along microtubule tractts toward the minus ends at the MTOC near the center of the cell 2. KINESIN motor -Disperse the melanosomes -Towards the periphery because the microtubule plus ends are orientated towards the periphery -Toward the periphery makes the skin cells appear darker |
|
|
How do cAMP concentrations relate to skin cells? |
-Decrease in cAMP then the melanin in the centre (Aggregated) -Increase in cAMP then the melanin is dispersed and chines pulls melanosomes to the plus ends |
|
|
How is cAMP a secondary messenger in skin cells? |
Signalling cAMP can control motor proteins and influence skin colour |
|
|
What were the first two evidences of protein transport into the nucleus? |
John Gurdon: Injection of histone proteins into cytoplasm, move to the nucleus
William Bonner: Labelled nuclear proteins injected into cytoplasm, move to the nucleus |
|
|
Nuclear shuttling: import and export |
1. Nuclear transfer experiments -Grown in radioactive and nucleus removed and put in nonradioactive cell and observed dikaryon which means that there are two nuclei so proteins move out of one nuclei and into another
2. Heterokaryon shuttling assay -Cytoplasm fusion of two completely different cells and a shuffling protein will relocate to the murine nucleus |
|
|
Nuceloplasmin |
-Regulates chromosome structure -Nuclear chaperone -Homo-pentameric complex -5 subunits
|
|
|
EXPERIMENT: First evidence of nuclear localization signal |
Three groups (radioactive heads, tails ad radioactive nucleoplasmin)
Injected into cytosol
Found it was in nucleus when just tails and nucleoplasmin |
|
|
How did that experiment test for sufficiency of signal? |
Non-protein components coated with tail proteins were injected into the cytosol and they accumulated at the nuclear envelope but were taken into the pores |
|
|
Is there a limit on cargo size? How can you test this? |
--Vary tail sizes to see which sizes get it and which ones get stuck |
|
|
What were the results of this? |
50 nm are lined up along the membrane half-way through the pore 50 nm appears to be the limit at which it can't get through the pore (threshold)
|
|
|
SV40 |
-Large T-antigen which induces production of antibodies -Required for viral genome replication |
|
|
How does SV40 get into nucleus? |
Can hijack cells nuclear import machinery to get into the nucleus
|
|
|
SV40 is it sufficient? |
Yes, because tagged with GFP and pyruvate kinase and it traveled to nucelus on its own |
|
|
SV40 is it necessary? |
Yes |
|
|
What is the t-antigen sequence? |
Proline-lysine x3 arginine lysine valine |
|
|
Explain the import machinery of RAN GDP |
-Diffuses through nuclear pore, no transport needed |
|
|
Explain the import machinery of a protein w/ nls |
Brought in my importin -RAN-GTP is used to take apart importing and protein -Importin alpha stays in the nucleus and RAN exports importing beta out -RAN-GDP dissociates the complex |
|
|
Influenza A virus and how does it enter? |
-The genome eight single stranded, negative-sense or anti-sense RNA molecules which are complementary to mRNA
-Inject 8 genome into cell |
|
|
vRNPs |
The vRNPs are the entities that enter the nucleus, and their nuclear import must be mediated by nuclear localization sequences (NLSs) exposed on the vRNPs. NP contains at least two putative NLSs, one at the N-terminus (NLS1) and one in the middle (NLS2) of the protein. These NP NLSs have been shown to mediate the nuclear import of recombinant NP molecules. However, it remains to be determined which NLS mediates the nuclear import of influenza vRNP complexes. |
|
|
Points of influenza |
-Inhibition of either NLS1 or NLS2 on NP proteins reduces import of nRNP (drop in infection) -Inhibition of NLS1 and NLS2 has an additive effect so both are important for import of vRNP and viral infection -Development of antiviral therapeutics will require targeting both NLS1 and NLS2 |
|
|
What do secondary messengers do? |
cAMP activate PKA cGMP activate PKG DAG activates PKC IP3 opens Ca 2+ channels in ER |
|
|
What happens when you bind an agonist to GPCR? |
-Leads to an activation of heterotrimeric G proteins |
|
|
How are GPCRs internalized for degradation? |
-GRK binds and removes g protein and arrestin interacts with AP2 adaptor to form endosome and targeted for degradation |
|
|
Nephron |
-Receptor in kidney that regulates water |
|
|
Explain osmoregulation at the kidney |
-Descending limb is permeable to water (90%) --Primary urine decrease and becomes more concentrated (looses water)
-Ascending limb is impermeable to water --Primary urine becomes more concentrated
-Collecting duct uses established gradient to produce urine of varing concentrations --Based on hydration |
|
|
Osmolarity of blood is under feedback by |
ADH on the collecting duct --Counteracts low blood pressure and high plasma osmolarity |
|
|
Explain the process of ADH |

|
|
|
What is needed for water permeability? |
Basolateral ADH receptor & luminal aquaporin 2 |
|
|
Explain what happens in the collecting duct and how concentrations are changed |
ADH activates adenylate cyclase by GFCR which uses ATP to produce cAMP -cAMP activates PKA and lets more fusion of AQP-2 to reduce the rate of endocytosis -More receptors on surface allows for more water to be let in from the apical side/urine passing side and more water in medulla which will then help with low blood pressure etc |
|
|
Congenital nephrogenic diabetes insipidus |
-Mutations in ADH receptor -Excessive thirst and hypotonic urine -Dehydration, fatigue and seizures due to electrolyte imbalance along with enlarged bladder |
|
|
Defects of GPCRs |
Class I -No transcription
Class 2 -Improper translation retention in the ER -Receptors are folded in ER so Congenital nephrogenic diabetes insipidus is class 2 defect
Class 3 -Retention in the golgi
Class 4 -Altered ligand binding, signal transduction and internalization
|
|
|
What is the cause of Congenital nephrogenic diabetes insipidus? |
Inactivating mutations --Impairment receptor function
--Can be 221 known ADH receptor mutations --Misfolded and trapped in endoplasmic reticulum --No AQP 2 expression or transport to luminal membrane |
|
|
Result of Congenital nephrogenic diabetes insipidus |
-Lack of receptors on basolateral membrane and lack of aquaporin channels on luminal membrane of collecting duct |
|
|
Pharmacochaperones |
-Diffuse into the cell, bind misfiled proteins and influence folding |
|
|
HEK treatment |
Expressing ADH receptor mutant with pharmacological chaperone could rescue receptors by promoting proper folding, increased localization at the membrane and responsiveness to ADH |
|
|
Observed results of pharmacological chaperones |
-Decrease in urine volume -Decrease in water intake -Increase in urine osmolality -Constant blood plasma osmolality |