Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
212 Cards in this Set
- Front
- Back
|
5 fingers of clinical diagnosis
|
History:
- chest pain/discomfort, dyspnea, palpitations, syncope, ankle edema (+/- orthopnea, paroxysmal nocturnal dyspnea, acute pulmonary edema, fatigue, cyanosis, hemotypsis, fever) - History of: medications, rheumatic fever, heart murmur, pregnancy complications, hypertension Physical: general appearance (Down's, Marfan, cyanosis), arterial pulse, venous pulse, precordial movements (apical impulse, parasternal lift) ECG/Xray: exercise stress testing, Holter/event monitor Non-invasive studies: ankle-brachial index, ECHO, nuclear stress testing, other imaging Invasive Procedures: cardiac cath, angiography, CT, intravascular ultrasound |
|
|
5 fingers of physical signs
|
General appearance:
Arterial pulse: evaluates left heart Venous pulse: evaluates right heart, wave form Precordial movement: apex (5th ICS, mid clavicular, for LV), parasternal lift (for RV) Auscultation: aortic, pulmonic, tricuspid, mitral |
|
|
5 fingers of the Cardiovascular evaluation
|
...after discovering a problem from your exam/workup:
1. Evaluate each lesion w/ regards to age, sex (+/- pregnancy), and race 2. Establish a diangosis 3. Determine the hemodynamic effect: effect on the heart or circulation, pathological/physiological consequences 4. Estimate severity: guides treatment choices (evaluate based on physiologic changes due to disease progression) 5. Treat appropriately: do no harm |
|
|
4 phase of diastole
|
- isovolumic relaxation: decrease in LV pressure between Ao valve closure and Mi valve opening
- early passive filling: (most of filling occurs, 2/3) - diastasis phase (plateau) - late diastolic/atrial kick: 20-25% of filling volume |
|
|
Cardiac pressures and O2 saturation
|
SVC (68% O2), IVC (71% O2)
RA (75%, 2-7mmHg) RV (75%, 15-30/4 mmHg) PA (75%, 15-30/8 mmHg PCap (97%, 12 mmHg) LA (95%, 2-10mmHg) LV (95%, 100-140/ 3-12 mm Hg) Ao (95%, 100-140/80) |
|
|
MI chest pain presentation
|
Onset: sudden
Quality: +/- pain, squeezing, tightness, heartburn, weight on chest, lump in the throat general discomfort (or none). Location: center of chest, diffuse pain Radiation: shoulders, arms (left arm is classic for coronary ischemia), neck, throat, lower jaw, teeh - Duration: persistent - Provocation: exertion, cold emotional stress, meals - Alleviation: nitroglycerine, cessation of activity |
|
|
Angina pectoris pain presentation
|
Onset: sudden, during activity
Quality: variable: numbness, burning, squeezing, tightness, choking, indigestion, pain, pressure Location: left of mid-sternum Radiation: left shoulder and upper arm, medial aspect of arm, elbow, forearm, wrist, shoulder or arm, lower jaw, nape of neck, interscapular area Duration: <3min after cessation of activity, 20min if after heavy meal, >30 if unstable angina Provocation: exertional or emotional stress Alleviation: Rest, nitroglycerine, staying upright |
|
|
Aortic dissection pain presentation
|
Onset: abrupt, pain greatest at onset
Quality: sharp tearing, painless dissection (6.4%) Location: posterior or anterior chest (descending vs ascending aorta), back Radiation: anywhere in thorax or abdomen Duration: persistent Provocation: spontaneous or from repetitive torque Alleviation: morphine, blood pressure control (reduce to normal systolic: <100mmHg, HR<60bpm) |
|
|
Pericarditis pain presentation
|
Onset: gradual
Quality: severe, sharp, pleuritic (variable depending on condition) Location: retrosternal, left precordial, may be pleuritic Radiation: trapezius ridge, neck, arms, left shoulder Duration: persistent Provocation: sitting up, inspiration, coughing Alleviation: lying down, leaning forward (reduce pressure on parietal pericardium) |
|
|
Acute coronary syndromes women vs. men
|
- women are more likely to have unstable angina than MI
- more likely to delay seeking emergency care - usually present older - tend to have more complications - 42% don't experience classic MI symptomatology |
|
|
Tachycardia
|
Definition: HR > 100 bpm
Types: sinus, atrial, ventricular Pathophysiology: shortens diastole, reduces LV filling, reduces coronary artery filling (so always slow HR after MI) |
|
|
Atrial Fibrillation
|
= temporally and spatially varying rapid, disorganized atrial electrical activation and uncoordinated atrial contraction
- no normal atrial activity to no atrial kick: lose - 20-25% of ventricular filling. - may be paroxymal or chronic Beside correlate: pulse deficit (not every beat gets to periphery), variable S1 intensity Etiology: - atrial dilation (RA/LA) causing it's conduction system to go faster than SA node - SA node inhibition: often due to infarct - hyperthyroidism Complications: - Thrombotic events: decrease in effective contraction results in hemostasis an large thrombus formation, potential embolus to major organs (incl. brain) - hemodynamic deterioration: loss of AV synchrony causing decreased CO, rapid/irregular ventricular rhythm reducing CO as well |
|
|
Radiological shadows of the heart and great vessels
|
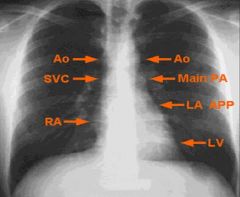
|
|
|
Key differences between fetal and adult circulation
|
- Source of oxygenation: lungs vs placenta (fetal umbilical arteries, from the internal iliac artery, are DEOXYGENATED)
- Ductus venosus: shunts blood from the umbilical vein to the inferior vena cava bypassing the liver - Foramen ovale: shunts oxygenated blood from the right to the left atria bypassing the lungs (angle of IVC directs blood right into foramen) - Ductus arteriosus: shunts blood from the pulmonary artery (mostly deoxy from the SVC) to the aortic arch bypassing the lungs |
|
|
Early morphogenesis of the heart
|
Fusion: heart tube is formed by day 22 by the fusion of the paired heart tubes. Midline heart tube has 3 layers (endothelial, cardiac jelly, splanchnic mesoderm) and is continuous w/ the aortic arches rostrally and venous system caudally
Formation of heart loop: enlongation and alternate dilation/constrictions form primative chambers: truncus arteriosus, bulbus cordis, primitive ventricle, primitive atrium, sinus venosus. Folding and rotation position the atrium/sinus venosus above and behind the other stuff by day 28. Septation: occurs between 4-6wks. Atrial: septum primum then secundum descend to the endocardial cushion leaving the ostium primum/secundum/then foramen ovale open. Ventricular: primitive interventricular septum grows up leaving interventricular foramen until wk 7 when membranous portion forms. Venticular outflow tracts also separated by bulbar ridges Development of cardiac valves: occurs via growth/apoptosis |
|
|
Septation of the heart
|
Atrial:
- septum primum descends superiorly and fuses w/ the endocardial cushion (closing the ostium primum). Ostium secundum then forms via resportion to allow for shunting (abnormal resorption causes ASD) - more muscular septum secundum then grows right, superiorly leaving open the foramen ovale. - Septum primum then acts as a flap to allow unidirectional shunting before birth Ventricular: - muscular ventricular septum grows from the inferior portion leaving the intraventricular foramen - the endocardial cushions then grow down forming the membranous portion (most common place for VSD) A-V: - endocardial cushions protrude into the AV canal, act as primitive valves - anterior and posterior cushions then fuse at the midline - valves then develop from local proliferations of tissue that reshape into leaflets Outflow tract: - bulbar ridges from in the truncus ateriosus and bulbus cordis. These ridges are offest in different segments creating a spiraling aorticopulmonary septum (failed spiraling leads to transposition of the great arteries). |
|
|
Situs relationships
|
Situs solitus = normal
Situs inversus w/ dextrocardia (situs inversus totalis) = complete mirror image of body organs, etc. usually benign Situs solitus with dextrocardia (due to L-looping) = normal organs, flipped heart, usually results in great vessel transposition Situs inversus with levocardia (situs inversus incompletus) = mirror image organs, normal heart. usually associate with great vessel transposition |
|
|
Cardiovascular changes that occur at birth
|
Shift from high PVR/R-to-L shunts to LOW PVR and increasing SVR, increasing pulmonary blood flow and closing the shunts (w/in 48 hrs). LV hypertrophy and RV atrophy develop due to high SVR and low PVR
|
|
|
L-R shunts (O2 step-up, overload, findings)
|
ASD:
- RA O2 step up - RV volume overload - findings: fixed S2 split. If advanced: parasternal lift (RV hypertrophy), dia/systolic murmers VSD: - RV O2 step up - most often LV volume overload due to flow directly into pulmonary artery (unless in muscular portion, <20%, causing RVVO) - findings: holosystolic murmur at LLSE. If advanced: apex displacement (LV hypertrophy), diastolic rumble Patent ductus arteriosus - Pulmonary artery O2 step up (return from Ao to PA) - LV volume overload - findings: continuous murmur at ULSE. If severe: apex displacement, diastolic rumble - can be associated w/ maternal rubella, altitude, premature birth. |
|
|
Aortic Coarctation findings
|
genetics: turner syndrome (XO)
Pathophysiology: LV pressure overload (due to high resistance in aorta) Exam findings: radial-femoral pulse delay If severe: LV hypertrophy causing apex changes, reduced leg blood pressure History: arm hypertension, leg fatigue, heart failure, dissection, stroke, rib notching (hypertrophied intercostal arteries), bicuspid aortic valve |
|
|
Congenital vs. Acquired cyanotic heart lesions
|
cyanotic lesions are associated with R-L shunt, returning unoxygenated blood to circulation.
Congenital: teratology of fallot, TGA. Associated with cyanosis, hypoxemia, digital clubbing, polycythemia, pulmonary, hypertension, paradoxical embolus. Acquired: Eisenmenger syndrome: reversal of flow through an existing L-R shunt |
|
|
Teratology of fallot characteristics
|
1. VSD
2. pulmonic stenosis (determines hemodynamic effect/cyanosis) 3. dominant aorta 4. right ventricular hypertrophy |
|
|
Hemodynamic changes associated with pregnancy, labor/deliver, postpartum
|
Plasma volume: ↑ throughout pregnancy (↑↑plasma, ↑ RBCs, hemodilution, peak 28-34wks (net >1-1.5L, 30-50% ↑), sustained in L/D, declines PP
Stroke volume: ↑1st, remains high for 2nd/3rd, during L/D ↑15-25% (contractions ↑preload), ↑80% immediately PP (from placenta) declines over 3mo PVR: ↓during P due to ↑prostacyclin and NO (vessel dilation) and ↓responsiveness to NE/Angiotensin (vasoconstrictors), ↑ during L/D, returns to normal PP HR: gradual ↑10-20% over pregnancy, ↑ during L/D (stress, etc), ↓PP CO: ↑30-50% during P, ↑15, then 50% during L/D stages, ↑80% initially PP due to volume overload, normalized over 3mo BP: no change 1st, ↓2nd due to ↓PVR, ↑diastolic/systolic 3rd, normal L/D, normal PP CVP: Normal throughout all stages |
|
|
Hemostatic changes during pregnancy, labor/deliver, and postpartum
|
Protein S/C (coagulation cascade): ↓protein S and ↑resistance to active C (2nd/3rd), persists through L/D and 6wks PP
Clotting factor production: ↑ through P, persists in L/D and PP 6wks Platelet Activation: ↑ during P, persists in L/D and 6wks PP |
|
|
Effect of pregnancy hemodynamic changes on underlying cardiac problems
|
Obstructive (aortic or mitral stenosis): increase plasma volume and will result in increased volume overload/residual volume in the LV and LA, causing reduced flow to the placenta and potential LHF
L-R shunts or valvular regurgitation: will reduce CO, severe disease (RHF, arrhymias are more problem), usually treatment b4 P reduces risk Aortic disease (Marfan, Turner's): increased risk for dissection (due to aortic changes, ↑CO) Cardiomyopathies: patients w/ more severe disease (worse LV function or history of cardiac events) are at higher risk for heart failure/event due to increase CO/plasma volume, these women have decreased survival postpartum |
|
|
Maternal cardiac conditions contraindicated for pregnancy due to maternal risk
|
1) Pulmonary arterial hypertension
2) Eisenmenger syndrome 3) Marfan’s syndrome with aortic involvement (aorta diameter > 4cm) 4) NYHA Functional Class III of IV CHF 5) Cyanotic congenital heart disease 6) Severe left ventricular outflow tract obstruction |
|
|
4 main roles of the physician for caring for women of child bearing age with heart disease
|
1) Assess the risk posed by pregnancy to the fetus and to the mother and inform the patient accurately about the risk (e.g. – carefully distinguish between “You cannot have children because of your cardiac condition” and “You are fertile but should not get pregnant because of the risk to you / the fetus.”
2) Implement necessary medication changes if/when pregnancy is desired or detected to prevent teratogenesis 3) Develop a management plan for the pregnancy at each stage: term, labor and delivery and the post-partum period 4) Recognize possible genetic contributions to the patients heart disease and make referrals to the Genetics department when the patient is considering / is pregnant. |
|
|
Exogenous pathway of lipid metabolism
|
Dietary fat is emulsified in the intestines by secreted bile acids and taken up into chylomicrons (triglycerides, phospholipids, apolipoproteins, cholesterol) .
In capillaries lipoprotein lipase/Apo C-II cleaves free FA's for energy (muscle) or storage (adipose). Remnants then taken up by receptors in the liver (ApoE). Bile acids are return to the liver via the portal circulation |
|
|
Optimal lipoprotein levels
|
Total cholesterol: <200 mg/dL
LDL <100mg/dL HDL >40-45 mg/dL TG <150 mg/dL even lower is recommended based on risk factors |
|
|
Endogenous lipid metabolism pathway
|
- 2/3 of circulating cholesterol is synthesized in the liver
- synthetic pathways in the liver produce VLDL - LPL/ApoC cleave free FA's in the capillaries, remnant IDL (short lived) is either taken up by the liver or converted to LDL (most). - LDL circulates and is taken up by tissues or liver via LDL receptors - HDL synthesized in circulation, acts as cholesterol scavenger to return cholesterol from peripheral tissues to the liver |
|
|
Normal physiologic functions of lipoproteins
|
- transport of lipids, TAG and cholesterol in circulatory system
- supply energy to muscle (TAG) - provide cholesterol for synthesis of steroid hormones - precursors for cell membrane synthesis (cholesterol, TAG) - provide cholesterol for synthesis of bile acid - Apolipoproteins acts as ligands for cellular receptors and cofactors for various enzymes involved in lipid metabolism |
|
|
Maturation of the LDL receptor
|
- expression of LDLR on the surface of hepatocytes is transcriptionally controlled by tx-Factor SRE-BP (sterol response element binding protein)
- SREBP in ER lumen binds to chaperone SCAP modifying it so it travels to the golgi - in golgi it is cleaved, freeing transcription factor that travels to the nucleus binding SRE and activating LDLR expression. - this is regulated by intracellular sterols via domain on SCAP, which deactivates the protein in the presence of excess sterols. - increased expression of LDLR is generally protective as it removes LDL from circulation |
|
|
Drugs used to treat moderately elevated TAG (~500mg/dL)
|
- HMG-CoA reductase inhibitors (statins)
- Bile acid sequestrants (cholestyramine) - Cholesterol uptake inhibitor (Ezetimibe) - Combination statin/cholesterol uptake inhibitor (Vytorin) |
|
|
Drugs used to treat severely elevated TAG (>500mg/dL)
|
- Nicotinic acid (Niacin, B3)
- Fibrate derivatives (fenofibrate) - omega-3 polyunsaturated FA's (omacor) |
|
|
HMG CoA Reductase Inhibitors
|
- reversible competitive inhibitors of HMG-CoA reductase which catalyzes the rate limiting step (HMG-CoA→ Mevalonate) in cholesterol synthesis from FA in hepatocytes
- drugs end in "statin" - result in reduced LDL and TAG levels by forcing cells to use exogenous cholesterol (via SCAP/SREBP pathway activation) - 25-50% clearance of serum LDL, also elevate HDL to some extent. [TAG] <500mg/dL generally responsive. - first line therapy for most patients with elevated LDL - side effects: dose-related muscle complaints (myalgia, weakness, rhabdomyolysis), memory loss, DMII |
|
|
Bile acid sequestrants
|
- cholestyramine
- catalytic polymers bind negatively charge bile acids preventing reabsorption (also fat uptake), resulting in increased cholesterol excretion and reduced fat intake. - Increased bile acid synthesis results in up to 28% decrease in plasma LDL - Side effects: decreased absorption of drugs and fat soluble vitamins, poor taste |
|
|
Cholesterol uptake inhibitors
|
- Ezetimibe
- competitively inhibits the Neimann-Pick transporter (NPC1L1) which normally transports cholesterol from the intestinal lumen into the enterocytes. - results in up to 20% decrease in serum LDL |
|
|
Nicotinic Acid
|
- niacin (vitamin B3), indirectly lowers VLDL by 2 mechanisms
- non-competitive inhibitor of the final step of TAG synthesis (from diglycerol) in hepatocytes (DGAT2 enzyme), lowering VLDL in circulation - inhibits breakdown of HDL in the hepatocyte, increasing plasma levels. - results in ↓VLDL 45%, ↓LDL 20%, ↑HDL 30%, best results in severly elevated TAGs. >500mg/dL - side effects: intense flushing (due to PG production, relieve w/ NSAIDs), hyperuricemia, impaired insulin sensitivity |
|
|
Fibrate derivatives
|
- are ligands for nuclear receptor, PPARa. Binding results in altered expression of multiple genes in lipid metabolism and inflammatory response (depending on co-factors recruited to receptor complex: drug, PPARa, retinoid X)
- including reduced Apo C-III (LPL and hepatic lipase inhibitor), and increased Apo A-V (stimulate LPL) both causing ↑VLDL breakdown - results in ↓VLDL production in hepatocytes, ↑VLDL breakdown, ↑HDL production - overall can result in ↓TAG 50%, ↓LDL 15%, ↑HDL 20% |
|
|
Omega-3 Polyunsaturated Fatty Acids (O3PUFA)
|
- 2 Omacor (O-3 PUFA's), effect is dose dependent, up to ↓TG 40%
- ligand for nuclear receptor PPARa causing altered expression of genes related to lipid metabolism and inflammation (depending on cofactors recruited to the complex) - Results in inhibition of TAG synthesis and reduction on VLDL release in hepatocytes. |
|
|
Systemic arterial hypertension
|
- elevation of systolic blood pressure causing an increased risk of cardiovascular events and target organ damage (pulse pressure SBP-DBP is impt for determining risk; 5mmHg ↑SBP/DBP = ↑30% CV risk)
- affects 60-65 mil in US (2/3 uncontrolled) (higher in men, post-menopausal women, blacks, >55y BP correlates directly w/ mortality) - 94% cases are primary/essential, - caused by any factor that increase cardiac output and/or total peripheral resistance: obesity, aging, excess alcohol, excess Na+, low K+, low Ca++, excess angiotensin II/endothelin, insufficient NO, insulin resistance, sedentary lifestyle, hereditary predisposition - Secondary HTN (5-7%, often reversible if underlying condition treated): renal dysfunction, endocrine abnormalities, aortic coarctation, medications, obstructive sleep apnea - malignant if DBP >120mmHg (usually renal failure, acceleration of mild HTN). Rapid and progressive presents w/ acute PE, acute renal failure, encephelopathy (vision, stroke) |
|
|
Hypotension
|
- nebulous definition of low blood pressure, no set standards (? <80-90mmHg)
- treated when the patient becomes symptomatic or if there is a dramatic change from the normal |
|
|
Target organs effected by HTN
|
- Heart (LV hypertrophy, CAD, HF, AFib)
- Vessels (aortic dissection) - Kidney (Chronic disease) - Brain (stroke) - Eye (hemorrhage) |
|
|
Reflex control of blood pressure
|
Standing causes venous pooling (↓preload) → transient ↓BP (alternative caused by ↓Na, after meal, emotional stress)
results in reflex action to correct BP → Baroreceptors fire (carotid, aortic) → ↑adrenergic tone (↑sympathetics, ↓parasympathetics) → vasoconstiction and ↑HR/LV contractility → BP returns to normal |
|
|
Blood pressure abnormalities
|
- Systemic arterial hypertension: primary/secondary, malignant, systolic only
- hypotension - abnormal pulse pressure: (SBP-DBP) no set standards, just if abnormally wide (high CO) or narrow (CO is low) - paradoxical pulse: large decrease in SBP (>10mmHg), often associated with cardiac tamponde, constrictive pericarditis, heart failure, asthma, OSA - Arm difference (>10mmHg) or Arm/leg difference: indictes damage to vessels (aortic coarctation) |
|
|
LaPlace Law
|
Tension = Pressure X Radius / Wall Thickness
Will predict how the ventricle responds to Pressure Overload (concentric hypertrophy) of increased tension (volume overload leads to dilation) |
|
|
Sequelae of LVH
|
- dystolic dysfunction (trouble getting blood into the cavity): due to ↓LV volume, ↓compliance, ↓wall relaxation resulting in ↓passive filling. Must rely on atrial kick to fill, causing atrial dilation (due to remaining volume), pulmonary edema (back flow), hemostasis (clotting), AFib (dilation pulls apart SA node tracts)
- MI: due to ↓coronary blood flow - eventually systolic dysfunction due to ↓CO from decreased filling and loss of LV contractility |
|
|
Arteriosclerosis
|
= generalized hardening of the arteries (loss of elasticity), usually of medium to larger arteries
- Can result from 3 vascular diseases: medial calcific sclerosis, arteriolosclerosis, actherosclerosis |
|
|
Mönckeberg medial calcific sclerosis
|
= deposition of calcium in the tunica media (musclar part) of arteries over time (presents in patients in 40-50's)
- does not cause narrowing or ischemia but may cause intermittent episodes of hypertension. - may coexist with atherosclerosis - radial artery will feel hard on physical exam |
|
|
Arteriolosclerosis
|
= hardening/loss of elasticity in the small arteries and arterioles, particularly of the kidneys
- often occurs due to long term HTN or DM - causes thickening of the arteriole walls and lumen narrowing, potentially leading to ischemia - |
|
|
Atherosclerosis
|
= accumulation of lipids, inflammatory cells (esp. monocytes) and extracellular matrix (collagen) within the intima of arteries (may involve muscular layers over time)
- involves large elastic arteries in systemic circulation and muscular arteries, paricularly the coronary, internal carotid, cerebral, and lower extremity - bifrucations, bends, and branch points (esp iliac artery bifrucation off aorta) are partcularly prone to plaque deposition due to turbulence of flow. Lower extremities tend to be more effected than upper. - 3 major sequelae: stenosis of muscular arteries (causing ischemia), thombogenesis (from ruptured plaques), aneurysm formation (loss of vessel recoil/elasticity, wall weakening) |
|
|
Risk factors for atherosclerosis
|
Overall: risk factors lead to endothelial dysfunction, characterized by ↑permeability (to lipids & inflamm. cells), ↓NO synthesis/release, ↑ROS, ↑adhesion molecules, ↑chemoattractants. Results in activation of SM, monocytes, fibrinogenesis
1. Hypercholesterolemia: oxidized lipids induce oxidative stress by reducing glutathione, resulting in ROS generation and endothelial dysfunctoin 2. Hypertension: elevated BP ↓ NO release,↑vascular permeability, ↑leukocyte adherence 3. Smoking: ↑platelet activation, ↑fibrinogen concentration, ↑monocyte adhesion, ↓HDL 4. Diabetes: induces hypercholesterolemia, associated w/ HTN, causes changes in LDL (excessive glycosylation) effecting turnover and depostion, ↓HDL 5. Localized flow abnormalities: ↓shear force so ↓NO release, ↑stasis so ↑thrombogenesis |
|
|
Development of atherosclerosis
|
In at risk patients lipids and cholesterol from LDLs travel through the endothelium, becomes oxidized by ROS and other factors. Oxidized LDL is associated w/ ↑expression of leukocyte adhesion molecules (ex: VCAM-1) attracting circulating moncytes to subendothelial tissue where they are converted to macrophages (by M-CSF). These then phagocytose lipids (oxidized kind is unregulated), becoming foam cells.
As disease progresses, foam cells accumulate, macrophages die releasing fibrogenic growth factors that activate SM cells to secrete ECM. Lipid remnants exist as extra-cellular crystallized cholesterol. Cytokines, extracellular collagen, and lymphocytes releasing cytokines together activate SM to move up to the intima and secrete collagen forming the cap of the atherosclerotic plaque. |
|
|
Fatty streaks
|
= early lesions of atherosclerosis consisting of foamy macrophages +/- additional T lymphocytes, aggregated platelets, or localized smooth muscle cells in the intimal layer
- seen in almost all children over age 10, may or may not progress to plaques - appear as irregular off white to yellow-white discolorations, non-palpable, near the luminal surface of the artery |
|
|
Atherosclerotic lesions/plaques
|
Stable:
- may or may not be symptomatic - rich in extracellular matrix and SM cells (thick capsule protecting lipid core) Unstable: - rich in macrophages and foam cells, prone to progressive growth w/o increasing stability - thin, weak fibrous cap over a large lipid deposit, making it prone to rupturee - tend to be eccentric (arising from the side of the vessel wall) Complicated: - plaque cap erosions or rupture, intra-plaque hemorrhage or superimposed thrombosis - risk factors: erosion or rupture of the plaque, mural thrombus, vessel weakening causing aneurysm. - may cause ischemia and AMI/angina prectoris, stroke, PVD, hemaotoma, ruptured anerysm |
|
|
Factors contributing to the disruption of atherosclerotic plaques and thrombi
|
- large extracellular lipid content
- thin fibrous cap - high content of inflammatory cells (lymphocytes and macrophages) producing proteolytic enzymes |
|
|
Development of atherosclerosis
|
In at risk patients lipids and cholesterol from LDLs travel through the endothelium, becomes oxidized by ROS and other factors. Oxidized LDL is associated w/ ↑expression of leukocyte adhesion molecules (ex: VCAM-1) attracting circulating moncytes to subendothelial tissue where they are converted to macrophages (by M-CSF). These then phagocytose lipids (oxidized kind is unregulated), becoming foam cells.
As disease progresses, foam cells accumulate, macrophages die releasing fibrogenic growth factors that activate SM cells to secrete ECM. Lipid remnants exist as extra-cellular crystallized cholesterol. Cytokines, extracellular collagen, and lymphocytes releasing cytokines together activate SM to move up to the intima and secrete collagen forming the cap of the atherosclerotic plaque. |
|
|
Fatty streaks
|
= early lesions of atherosclerosis consisting of foamy macrophages +/- additional T lymphocytes, aggregated platelets, or localized smooth muscle cells in the intimal layer
- seen in almost all children over age 10, may or may not progress to plaques - appear as irregular off white to yellow-white discolorations, non-palpable, near the luminal surface of the artery |
|
|
Atherosclerotic lesions/plaques
|
Stable:
- may or may not be symptomatic - rich in extracellular matrix and SM cells (thick capsule protecting lipid core) Unstable: - rich in macrophages and foam cells, prone to progressive growth w/o increasing stability - thin, weak fibrous cap over a large lipid deposit, making it prone to rupturee - tend to be eccentric (arising from the side of the vessel wall) Complicated: - plaque cap erosions or rupture, intra-plaque hemorrhage or superimposed thrombosis - risk factors: erosion or rupture of the plaque, mural thrombus, vessel weakening causing aneurysm. - may cause ischemia and AMI/angina prectoris, stroke, PVD, hemaotoma, ruptured anerysm |
|
|
Factors contributing to the disruption of atherosclerotic plaques and thrombi
|
- large extracellular lipid content
- thin fibrous cap - high content of inflammatory cells (lymphocytes and macrophages) producing proteolytic enzymes |
|
|
Important questions to ask for suspected angina pectoris
|
- Quality: chest discomfort, pain, squeezing, tightness, "elephant on chest"
- Location and radiation: "dinner plate size", central, radiations to jaw, back, arms - Provocative and palliative factors: exercise worsens (increase O2 demand of the heart) - Duration: should last more than 30-60sec. Angina is lactic acidosis buildup, which takes a few minutes to dissipate - Associated symptoms: arm pain, jaw pain, exertional discomfort, pain between shoulder blades etc - PMH/PSH: prior CAD or cardiac testing, smoking, diabetes, HTN, lipids - FH: relatives with CAD, MI, smoking lessens genetic risk |
|
|
4 classes of chest pain by history
|
- Definite Angina: classic symptoms predictably provoked by exercise, relieved by rest. Discomfort may radiate to back, arm, jaw
- Probable angina: mostly classic symptoms with some variation. Ex: some chest pain but varying quality (sharp vs dull) - Probably not angina: atypical symptoms that do not totally exclude ischemia - Definitely not angina: if sharp twinge of pain is felt or if pain lasts <30-60s |
|
|
5 classes of angina
|
Class 0: no angina
Class I: Angina only with very strenuous exertion (running) Class II: Angina only with walking or climbing hills or stairs rapidly or >2blocks rapidly on ground Class III: angina on 1 flight of stairs or <2 blocks on level ground Class IV: angina with minimal exertion or at rest |
|
|
Chronic Stable Coronary Disease definition
|
- coronary disease involving at least one hemodynamically significant lesion
- may either be asymptomatic ischemia on stress test or angina with exertion - disease is stable/consistent with regards to the amount of exercise needed to provoke symptoms as well as the duration and severity when they do occur |
|
|
Types of therapies for chronic stable CAD
|
- life style modification: most neglected, hardest to achieve
- pharmacologic therapy - percutaneous coronary intervention - coronary artery surgery Goals of all therapy: prolongation of life, relief of symptoms |
|
|
Benefits/limitations of percutaneous coronary intervention (PCI)
|
Benfits:
- reduction in symptoms due to widening of arteries--improve flow Limitations - only treating a small portion of the coronary tree (50 cm vs 1-2cm stent) - limited impact on mortality (most MI's don't occur in heavily narrowed plaques) |
|
|
Results of the COURAGE trial
|
stenting stable angina = "whack a plaque"
- 2287 patients w/ stable CAD randomized to medical tx or PCI, followed for 4.6 years. A few exclusions Results: - no overall difference in mortality or MI/ACS free survival between medical therapy and PCI - BARI 2D Trial: in diabetics showed revasculariztion had increased event free survival compared to medication but no change in mortality - |
|
|
3 main categories of medication for reduction of MI and death
|
Aspirin:
- reduces incidence of vascular events by 20-25% in pts w/ prior events or otherwise at risk Beta Blockers: - after MI reduce death and recurrent MI 20-30% Statins: - lower mortality 20-30% in known/highly suspected CAD |
|
|
Indications for PCI vs CABG in stable patients
|
PCI
- intolerable angina despite maximal Rx in a patient who does not require CABG for prolongation of life - Markedly early positive stress test without absolute indication for CABG (group not included in COURAGE trial) - Older patients who are at very high risk for CABG (stenting possible safer?) CABG - left main coronary narrowing (>50%) - 3-vessel CAD (esp with poor LV function). Some of these patients can still be stented or managed medically |
|
|
Lifestyle modications that are helpful in chronic stable CAD
|
- BP control
- smoking cessation - weight reduction - daily exercise - optimize lipid (diet, pharm) |
|
|
3 classes of drugs for reducing symptoms of chronic angina
|
Overall mechanism: either ↓O2 demand or ↑O2 supply
Beta blockers: first line - blockade of Beta-1 receptors reduce O2 consumption by ↓HR, ↓contractility, ↓wall stress - adverse effects: bradycardia, bronchoconstriction (asthma), fatigue/depression, worsened PVD symptoms, erectile dysfunction Calcium Channel Inhibitors: (verapamil, diltiazem, dihydropyridines) - reduce O2 demand by blocking Ca++ entry into cells, decreasing vascular tone: vasodilation, ↓HR (some), ↓contractility (some), ↓ wall stress through ↓BP, no survival benefits Nitrates: - cause systemic vasodilation (↓wall stress) plus coronary vasodilation, no survival benefits - tolerance results from chronic use, sublingual and transdermal types Ranolazine: - reduces Ca++ in ischemic muscle and inhibits FA oxidation. - prolongs QT interval, contraindicated w/ liver disease, Ca++ blockers |
|
|
Ischemia associated heart diseases
|
>90% due to atherosclerotic CAD
Types: - coronary artery vasospasm (cocaine) - coronary artery emboli - hypotension/shock (insufficient flow to perfuse adequately) |
|
|
Types of angina pectoris
|
Stable angina:
- due to reduced perfustion of the myocardium due to chronic stenosis/obstruction. Tend to have ischemia on exertion Prinzmetal angina: - episodic angina at rest due to coronary vasospasm (esp cocaine users). Not associated with plaques or lead to associate thrombi Unstable angina: - progressively worsening chest pain with decreasing exertion. Plaques are at risk for rupture and thrombosis (usually small, not fully occlusive, but if large then MI) |
|
|
MI pathogenesis
|
1. disruption of a plaques exposing thrombogenic factors (collagen, Von Willebrand factor, fribronectin, etc)
2. Platelet adhesion and activation (granule release) 3. Activation of the coagulation pathway 4. Lumen occlusion and tissue ischemia |
|
|
Morphologic changes in the heart after MI
|
<12hrs: no noticeable macro or microscopic changes
24hrs: tissue looks mottled, dark red color due to coagulation necrosis, micro see loss of nuclei 1-3 days: gross yellowing surrounded by mottling, micro dead muscle tissue with neutrophilic invasion and expansion of surrounding vessels due to inflammatory cell recruitment 3-10 days: red border around yellow infarct due to accentuation of surrounding vessels. Micro see increase macrophages and tissue destruction (via macrophages and neutrophils) >10 days: yellow white scar, on micro see fibrous scar tissue |
|
|
Complications of MI
|
- contractile dysfunction
- conduction abnormalities: especially if infarct occurs along conduction bundles - arrythmias: can lead to mural thrombi - pericarditis - myocardial rupture: occurs mostly 4-7 days post MI, could be papillary or wall, often fatal (but it is uncommon) - ventricular aneurysm - progressive late heart failure: due to loss of adequate perfusion |
|
|
Function of endothelium
|
- barrier function: prevents anomalous material from adhering (anti-thrombotic)
- metabolically active exchange medium: transmits chemicals, material (proteins, lipids), and cells (leukocytes) between the lumen and submucosa - regulates blood flow: produces NO which induces vasodilation by relaxing SM, inhibits platelet aggregation, prevents mononuclear adherence/migration into perivascular tissue |
|
|
Dysfunction of epithelium in atherosclerosis
|
Dysfunctional endothelium promotes accumulation of lipids within the perivascular tissue, leading to cyclical phagocytosis and apoptosis of macrophages, increased expression of monocyte adhesion molecules (VCAM-1) on endothelial surface, and release of cytokines initiating the fibrotic pathway. These then lead to continued lipid accumulation, leukocyte infiltration, and SM activation
- endothelial dysfunction is the link between risk factors and the development of disease - particularly important is the loss of NO production (only in normal, metabolically active endothelial cells) - lipid hypothesis: lipids are transported across the endothelium but unlike normal are not transported back |
|
|
Development of vulnerable plaque
|
- intimal lesions grow outward first (positive remodeling) then a can grow into the lumen (negative remodeling)
- the rate at which the plaque is deposited contributes to vulnerability: if quickly deposited the lipid core is larger with a smaller fibrous tissue cap (less response time) making it more prone to rupture. Slower deposited plaques are interlaced with fibrous tissue making them more stable. - Over time vulnerable plaques can develop by accumulation of lipids thinning out the fibrous cap. |
|
|
Types of aortic dissection
|
Type 1:
- occurs proximal to the left subclavian artery in the ascending aorta - medical emergency requiring immediate surgical repair, use anti-HTN meds to control BP - w/o treatment 48% will die in 2 days. Type 2: - occurs distal to the LSCA in the descending aorta (thoracic or abdominal region) - treated medically rather than surgically using BP medications |
|
|
Important signs of aortic dissection
|
- difference in BP between the arms (type 1): due to different flow volumes to L/R SC's
- history of tearing chest pain through to the back with maximal intensity at onset, no radiation: due to physical tearing of aorta - decrease sensation or movement of parts of the body, shortness of breath, rapid or weak pulse, fainting, nausea and vomiting, changes in cognition - |
|
|
Major complications of arotic dissection
|
- aortic regurgitation: occurs acutely and severely due to dilation of the aorta compromising valve integrity (creates spaces between cusps)
- pericardial effusion: due to leakage of blood into the pericardial space causing tamponade - Death - ischemia in other parts of the body |
|
|
Physiological basis of angina
|
- angina pectoris is a symptom of ischemic heart disease caused by an imbalance between oxygen demand and supply by the coronaries.
- supply restriction is usually due to CAD (atherosclerotic plaques or coronary vasospasm). - Reduced blood flow to the muscle tissue results in build up of metabolites which stimulate nerves causing discomfort: lactic acid, histimine, K+, H+, adenosine |
|
|
Physological alterations that can relieve angina
|
Increase O2 supply: (not effectively increased in angina due to existing maximal dilation, restrictive plaques)
- vasodilation: adenosine (AMP/ADP) builds when ischemic (not converted to ATP), binds to SM or endothelial receptors in arterioles causing ↑Ca++ influx and vascular relaxation, ↓SVR, ↑blood flow - Other substances produced by endothlium can contribute to vasodilation: NO (also inhibits platelet aggregation), prostacyclin, endothelium-derived hyperpolarizing factor (EDHF) - ↑diastole: increases the time for perfusion of the coronary arteries (which are only filled on diastole b/c of collapse under systolic pressures) Reduction in O2 demand (more effective target for angina treatment) - strategy: reduce the stress on the heart to ↓metabolic needs during contraction: - ↓preload: ventricular stress before systole/ LVEDP/V. Decrease venous return via ↑RA pressure, ↑PVR - ↓afterload: tension on ventricle during systole. ↓PVR - ↓contractile force: ↓Preload - ↓HR: ↓ β1 stimulation and ↑muscarinic stimulation |
|
|
Nitrovasodilators
|
- glyceryl trinitrate (GTN, nitroglycerin), isosorbide mononitrate, isosorbide dinitrate
mechanism: undergo enzymatic bioactivation to NO which results in upregulation of cGMP causing reduction of intracellular Ca+ and SM relaxation. - preferentially dilate venous circulation (greater enzymatic capacity), decreasing venous return and preload. Therefore ↓ventricular wall stress, ↓mycocardial O2 demand. No change in coronary blood flow. - No inhibits platelet aggregation, may help in unstable angina. Administration: - sublingual (GTN, ISDN, avoids first pass metabolism, rapid effect), transdermal (prolongs onset and duration) - tolerance: develops w/ all nitrates, does dependent, disappears after 24hrs, use nitrate free periods >8hrs and least effective dose Adverse effects: (excessive vasodilation, ↓BP) - orthostatic hypertension, reflex tachycardia, ↑cardiac contractile force, severe throbbing headache, dizziness, cross potentiation w/ PDE-5 inhibitors (viagra) |
|
|
Calcium channel blockers
|
Mechanism: block L-type ("slow") Ca++ channels, ↓Ca+ influx into cardiac and arterial SM. Result in preferential ↑arterial vasodilation, ↓cardiac contractility (different selectivity for Ca++ channels in different tissue)
- Nifedipine: high selectivity for peripheral vasodilation (good for Prinzmental angina). Dilitiazem and verapmil have greater contractility and HR reduction effects Adverse effects: rapid ↓BP causes reflex ↑HR/contractility that aggravates angina; synergy with beta blockers causing excessive bradycardia, AV block, inotropic depression |
|
|
β1 adrenergic receptor antagonists
|
Mechanism: inhibit beta receptors on cardiac and vessel SM causing ↓HR, ↓contractility, ↓afterload (in HTN patients), ↓diastolic interval (↑coronary filling)
- non-selective: propanolol (not indicated for asthma), selective: metoprolol (Lopressor) Adverse effects: - B2: may exacerbate: bronchospastic disease, raynaud's syndrome, vasospastic angina - B1: may exascerbate heart failure, produce excessive bradycardia or AV block, synergize w/ non-dihydropyridine Ca++ blockers causing AV block & bradycardia, abrupt withdrawl may precipitate tachycardia, angina, arrhythmia |
|
|
Combination therapies for angina
|
Nitrovasodilators + Beta blockers:
- BB' prevent relfex tachycardia and ↑contractility provoked by nitrate HTN - Nitrates and BB reduce myocardial O2 demand by different mechanisms Ca++ blockers and beta blockers: - BB's prevent reflex tachycardia and ↑contractility induced by CaB's - BB and CaB's have different mechanisms - BBs and non-dihydropyridine CaBs work synergistically to ↓HR/contractility, allowing for lower doses of each to be used |
|
|
Ranolazine
|
Mechanism: inhibits late Na+ current
During ischemia delayed Na+ channel closing causes ↑[Na+], which overloads Na/Ca exchanger causing ↑[Ca++] resulting in electrical instability, mechanical dysfunction, abnormal contraction/relaxation, increased diastolic tension - alleviates angina w/o dependence on BP or HR reduction so has fewer side effects. Good for chronic angina but not ACS Adverse effects: prolongs QTc, CYP3A4 substrate (drug interactions) |
|
|
Vasculitis
|
= inflammatory damage to the blood vessels causing hemorrhage and ischemia distal to the lesion
Clinical features: - most vasculitis is secondary, primary causes are uncommon, etiology may not be determined - skin is the most common site, especially the legs. Usually will include non-blanching purpura, possibly necrotic/ulcerated areas due to ischemia, +/- pain - also: lung lesions (hemotypsis), GI (bloody stool, melena), renal (hematuria) Classification: usually based on type of vessel involved does not reflect pathogenesis or histopathology Diagnosis: look for anatomic sites involved, granulomatous inflammation, ANCA serology, concurrent autoimmune dz, medications/toxins, infections |
|
|
Giant cell arteritis
|
= vasculitis of medium to large vessels, usually temporal, occipital, facial artery distribution. Unknown pathogenesis
- Effects elderly patients, F:M 3:1 Symptoms: - Visual disturbances: opthalmic or retinal artery involvement, blindness can result - polymyalgia rheumatica: muscle stiffness/pain in neck, shoulder, UE, jaw - neurologic signs/symptoms Lab: elevated ESR and CRP Path: - focal lesions, so false-negative biopsies are common - granulomatous inflammation of the artery wall, destruction of the internal elastic lamina, narrowing/occlusion of the vascular lumen (would have distal infarcts), neutrophilic infiltrate dominates |
|
|
Takayasu Arteritis
|
= vasculitis of the aorta and it's main branches (esp. subclavian artery)
- Epi: affects patients <40, F 7:1, rare in the US more common in Asia Clinical features: "pulseless disease," low blood pressure, ocular disturbances, carotid tenderness, unpredictable course (rapid progression, quiescent stages) Lab: elevated ESR Pathogenesis: not understood, diagnosis is clinical/radiological Histopath: granulomatous inflammation of the artery wll (can be identical to giant cell arteritis), destruction of the internal elastic lamina, narrowing/occlusion of the vascular lumen, neutrophils are dominant infiltrate |
|
|
Polyarteritis nodosa
|
= multisystem vasculitis (except lung) of medium sized vessels
- Epi: wide age range, M 4:1, strong association with Hep B - Clinical features: fever, weight loss, HTN, abdominal pain, melena, neuropathy, myalgias, purpuric/ulcerated skin lesions - Lab: elevated BUN or creatinine, Hep B antigen, ANCA negative - Hist: necrotizing vasculitis of medium-sized vessels, mimics other vasculitities |
|
|
Kawasaki disease (mucocutaneous lymph node syndrome)
|
= multisystem vasculitis effecting medium size vessels. May be infection related (staph)
- Epi: effects children and young adults, M>F Clinical features: erythematous conjunctive and oral mucosa/tongue, cervical lymphadenopathy, edema in hands and feet, desquamation of fingertips and toes - Cardiovascular involvement: 50% of patients (lethal in a small %), causes myocarditis (CHF), coronary artery vasculitis (ischemic disease, infarction, CA ANEURYSM formation--risk later in life) - Lab: ANCA negative, anti-endothelial antibodies |
|
|
Granulomatosis with polyangitis (Wegener's)
|
= multisystem necrotizing granulomatosis and vasculitis. Classic ELK involvement of systems
- Epi: middle aged patients, M 3:2 - Diverse sites involved: URT/sinonasal (saddle nose), pulmonary, renal, cutaneous, middle ear, peripheral nerves, CNS, ocular, oral cavaty Clinical presentation: - URT (sinusitis/rhinitis, destructive URT lesions), Lungs (cough, chest pain, hemoptysis, nodular infiltrates, cavitary lesions), Renal (hematuria, proteinuria) Lab: hematuria, red cell casts in urine, c-ANCA positive (70-80%) Histo: small vessel vasculitis (mostly lungs) with necrotizing granulomatosis |
|
|
Allergic granulomatosis with angiitis (Chrug-straus syndrome)
|
= multisystem vasculitis and granulosis
- Epi: affects young adults, M>F Clinical presentation: fever, cardiac involvement (arrhythmia, ventricular insufficiency, coronary arteritis, pericarditis/myocarditis), pulmonary infiltrates (patchy, nodular, non-cavitating), renal glomerulonephritis Lab: peripheral eosinophilia, elevated ESR, p-ANCA positive (60%) Histo: small vessel vasculitis with necrotizing granulomatous inflammation and abundant EOSINOPHILS, in skin involves dermis and subcutis |
|
|
Henoch-Schonlein Purpura
|
= multisystem small vessel vasculitis, possible infection of drug relatedd
- Epi: young children <10yrs, M>F - Clincial presentation: cutaneous purpura (leukocytoclastic vasculitis), abdominal pain (GI vasculitis, blood stools, melena), arthralgias, renal glomerulonephritis Lab: hematuria, proteinuria, ANCA negative Histo: leukocytoclastic vasculitis in superficial dermis (not specific), IgA deposition |
|
|
Microscopic polyangitis
|
= multisystem small vessel vasculitis, may be infection or drug associated
- Epis: affects adults - Clinical presentation: cutaneous purpura (leukocytoclastic vasculitis), abdominal pain (GI vasculitis, blood stools, melena), pulmonary (hemoptysis, intrapleural hemorrhage), renal glomerulonephritis, myalgias Lab: elevated ESR, anemia, hematuria, proteinuria, renal failure, p-ANCA positive (50-60%), c-ANCA positive (40%) Histo: leukocytoclastic vasculitis (not specific in isolation, requires clinical correlation) |
|
|
Key vasculitis associations
|
IgA vasculitis: Henoch-Schonlein
Absent clinical lung involvement: PAN Abundant eosinophils: Churg-Strauss Coronary aneurysms: Kawasaki dz. Giant cells: Giant cell and Takayasu Blindness: Giant cell Pulseless: Takayasu |
|
|
Chylomicrons
|
- Largest lipoprotein, lowest density
- Almost entirely lipids, and over 90% triglycerides - Deliver triglycerides (TAG) from the diet (exogenous) in the intestine to the adipose and muscle tissues - Chylomicron remnants (after the chylomicron has delivered most lipids) delivers the leftover lipids to the liver. This is mostly cholesterol and some leftover TAG |
|
|
VLDL
|
- Contain 60-70% triglycerides
- Almost entirely lipids, but higher protein content than chylomicrons - Produced by the liver - Biological function is to deliver endogenous triglycerides to adipose and muscle |
|
|
IDL
|
- Intermediate particle between metabolism of VLDL and conversion to LDL
- Action of receptors on IDL cause it to lose surface proteins and triglycerides - Becomes LDL after action of receptors |
|
|
LDL
|
- Major cholesterol ester carrying lipoprotein: 2/3-3/4 of serum cholesterol is carried in LDL particles
- Product of VLDL metabolism - 50% or particle mass is made up of cholesterol esters - Arises in the blood stream as VLDL is metabolized - LDL delivers cholesterol to many tissues |
|
|
HDL
|
- Smallest lipoprotein
- Synthesized by intestine and liver as a cholesterol-poor lipoprotein, then gains cholesterol and cholesterol esters from peripheral cells and other lipoproteins - Participates in reverse cholesterol transport: Removes excess cholesterol from cells in tissues, delivers cholesterol to the liver for metabolism (the liver is the only organ that can get rid of cholesterol) |
|
|
Lipoprotein structure
|
Very basic structure: hydrophobic lipid core, hydrophilic lipid surface monolayer
- Inner droplet of neutral lipids: triglycerides and cholesterol esters (cholesterol esters are always on the inside of the lipoprotein) - Soluble surface monolayer of phospholipids and unesterified cholesterol - Surface proteins (apolipoproteins) are attached to the outer lipid layer through their specific lipophilic domains |
|
|
Chylomicron metabolism
|
- Delivery of fatty acids to peripheral tissues is through the action of lipoprotein lipase (LPL)
- Interacts with circulating lipoproteins to break down triglycerides into free fatty acids - Adipocytes and muscle cells then take up free fatty acids - Apolipoprotein C-II (apoC-II – surface protein in chylomicron) activates LPL - Chylomicrons also lose extra lipoproteins during the removal of triglycerides, these extra lipoproteins go to HDL - Also lose some surface proteins, but apoB48 stays on surface - Becomes a remnant chylomicron particle with a slightly increased ratio of cholesterol to triglycerides - Chylomicron remnants are taken up by the apoB48 receptor in the liver |
|
|
VLDL metabolism
|
Delivers fatty acids that are made in the liver to the adipose/muscle
- Surface proteins apo C and apo E are acquired from HDL particles in circulation. Apo B and some apo E are present when the VLDL is made in the liver - VLDL also gets cholesterol esters from HDL, and becomes enriched in cholesterol ester and apolipoproteins - ApoCII activates LPL in capillary beds - VLDL delivers fatty acids to tissues, and releases apoE and apoC back to HDL - VLDL remnant is IDL, which can be converted to LDL or taken up by the liver and metabolized completely |
|
|
LDL metabolism
|
LDL is formed from IDL (VLDL remnants) and only has apoB-100 on surface
- Tissues get cholesterol from LDL because biosynthesis of cholesterol takes ~18ATPs - LDL delivers cholesterol to arterial wall where it is taken up by cells that recognize the apoB-100 on the surface of LDL: Receptor mediated uptake accounts for ~3/4 of uptake by the liver and for ~2/3 of uptake by other tissues - Nonreceptor mediated uptake of cholesterol is through pinocytosis or scavenger receptor mediated mechanisms |
|
|
HDL metabolism
|
Initial nascent HDL is a hollow shell. In circulation, starts to pick up cholesterol ester from other lipoproteins and peripheral tissues
- Peripheral tissues contain cholesterol transporters that allow for the efflux of cholesterol out of the tissue, HDL acts as a receptor - As cholesterol is being transported out of the tissue, it is esterified by Lecithin Cholesterol Acyltransferase (LCAT), which allows for the cholesterol to be stored in the core of the HDL particle - As cholesterol fills HDL, the particle grows larger - Some of the excess cholesterol can also be delivered to LDL or VLDL - HDL can deliver lipid to the liver, and then the liver can excrete the lipids as bile acids |
|
|
Treatment for familial hypercholesterolemia
|
- Combination lipid therapy to get cholesterol levels down to goal levels: statins, niacin, fibrates, cholesterol absorption inhibitors, bile acid sequestrants
- Homozygotes can sometimes show reduced response to statins - Surgeries are usually not indicated for treatment of FH itself: Portocaval shunting, Liver transplantation - Gene therapy has been ineffective because of mutations - LDL apheresis – similar to dialysis |
|
|
Functions of different parts of circulatory system in regulating blood pressure
|
Heart: pump, maintains BP via modulation of contractile force and HR
Kidney: volume regulator via salt/water control and renin/angiotensin system. Also has endocrine function, and Arterioles: major regulators of resistance due to high SM content, respond to distention with constriction, also have hormones, receptors, autacoids, nerves, etc that modulate BP Veins: capacitors: main storage of volume CNS: sympathetic regulation of tone |
|
|
Mechanisms of SM contraction and relaxation
|
Contraction:
- vasoconstrictors bind to cell surface receptors activating phospholipase C which leads downstream to the production of IP3, which acts on channels in the sarcoplasmic reticulum causing efflux of calcium - free Ca binds to myosin light chain kinase (MLCK) which phos's myosin chains allowing interaction with actin leading to contraction. - Free calcium also activates L-type Ca channels leading to influx of extracellular Ca to replenish sarcoplasmic reticulum Relaxation: - Beta-2 pathway: binding of NE to Beta-2 receptor leads to cAMP synthesis, which activates PKA and leads to relaxation (mechanism unknown) - NO pathway: NO is synthesized by endothelial cells in response to shear stress (Ca activates NOsynthase), NO regulates vascular tone, triggers cGMP production activating PKG - hyperpolarization pathway: K+ efflux causes hyperpolarization of SM cells, suppressing L-type Ca channels, reducing Ca+ in sarcoplasmic reticulum and therefore contraction of SM |
|
|
Minoxidil, hydralazine, diazoxide
|
= Potassium channel openers
Mechanism: (indirect Ca+ channel inhibition) targets K+/ATP channels opening them, leading to K+ efflux and cell hyperpolarization, inhibiting L-type Ca++ channels which reduces Ca+ and causes SM relaxation Main uses: HTN not controlled on other drugs, HTN emergency, severe HTN, research Adverse effects: strong induction of reflex to vasodilation: baroreflex stimulation of sympathetic NS → ↑volume retention and HR/contractility → increase myocardial ischemia |
|
|
Calcium channel blockers
|
- dihydropyridines, diltiazem, verapamil
Mechanism: selectively inhibit L-type Ca+ channels reducing intracellular Ca+ and muscle contraction (in both vascular and cardiac muscle tissue) - Dihydropyridines are more selective for vascular receptors and so have less effect on the heart - Indications: mild to moderate HTN for those patients that have associated ischemia |
|
|
Nitrates
|
- sodium nitroprusside, nitroglycerin
Mechanism: non-selective vasodilators that act as NO donors (bypasses endothelial mechanism of NO release). Results in vasoldilaiton, ↓pre/afterload ↓cardiac stress - Effect is primarily on veins because drugs are better activated there. - Indications: HTN emergencies, to reduce bleeding during surgery (diversion of blood to veins), patients with cardiac ischemia |
|
|
Valvular aortic stenosis physical exam findings (UMedic) (severe case)
|
History: possible murmur, exertional syncope, no family or personal history of cardiac disease
General Appearance: normal BP: low systolic, normal diastolic Carotid pulse: hypokinetic (decreased stroke volume due to decreased preload, systolic dysfunction, or outflow obstruction) Peripheral pulses: reduced (suggest obstruction) JVP: Normal Venous wave form: normal (dominant a, smaller v) Precordial movements: normal Apex:normally placed, enlarged (LVH), impulse is sustained + presystolic (LVH + decrease compliance) Auscultation: (URSB) long, late peaking, crescendo-decrescendo murmur with ejection sound (aortic) w/o respiratory variation, murmur radiates over carotids R>L; at apex S4 + S1 + ES Breath sounds: normal EKG: incease QRS voltage (LVH), ST-T abnomalities Treatment: balloon valvuloplasty or replacement |
|
|
Dilated cardiomyopathy physical exam (umedic)
|
History: dyspnea on exertion/at rest, peripheral edema, enlarged heart, paroxysmal nocturnal dyspnea, orthopnea, enlarged heart (no other personal/family history)
BP: diastolic borderline elevated, narrow pulse pressure Carotid pulse: hypokinetic JVP: elevated CVP; giant a smaller v (RVH) Abdomnojuglar test: (compress liver to increase return) abn:rise in CVP during compression and fall of 4cm after Precordial movement: LLSB "triple ripple": presystolic + systolic + early diastolic impulses (RVD/H, S3/S4) Apex: inferolaterally displaced (LVD), very enlarged; impulse is sustained + presystolic + early diastolic component Auscultation: ULSB split S2 on inspiration, S2 is louder; LLSB S3 + S4 on inspiration, Apex: holosystolic, plateau murmur (mitral regurg), S3 + S4 (LVD) Breath sounds: absent in LRP (pleural effusion from CHF), LLP (inspiratory crackles, pulmonary congestion) |
|
|
Hypertrophic cardiomyopathy
|
History: angina, syncope, dyspnea
Exam: rapid rising carotid pulse, hyperdynamic apex, S4 + systolic murmur Characterized by distolic dysfunction/reduced filling |
|
|
Restrictive cardiomyopathy
|
Characterized by dystolic dysfunction (walls impeded filling)
- simulates constrictive pericarditis - idiopathic or infiltrative (amyloidosis) Symptoms: dyspnea, fatigue Exam: elevated CVP, prominent X and Y descents |
|
|
Angina pectoris clinical presentation (Umedic)
|
History: lower chest discomfort, lasting minutes, precipitated by exertion/eating/emotion relieved by rest, avoiding heavy exertion, jaw/arm radiation, family/personal history of CAD risk factors
Possible clinical findings (often not many): obesity, HTN, xanthelasma/corneal arcus (hyperlipidemia), ectopic impulse on chest wall (ischemic bulge of myocaridum), paradoxic S2 splitting, S4 at apex, murmur of papillary dysfunction, during attack sustained systolic ectopic pulse Usually normal findings (abnormal could be an unusual cause of CAD): carotid pulse (abn. could be due to LV outflow track obstruction), peripheral pulses (no PVD), JVP, jugular wave form, apex location, apical impulse (may have pre-systolic and early systolic components), heart sounds, lung sounds |
|
|
HTN clinical presentation (UMedic)
|
History: dyspnea on exertion, moderate obesity, headaches, not other personal history, family history of HTN
BP: elevated Fundoscopic exam: arteriolar narrowing/spasm, AV nicking (grade IV would have papilledema, exudates, hemorrhages, arterial spasm) Pulses: normal JVD: normal Precordial movement: normal, apical impulse normally located but enlarged (LVH), impulse is sustained + presystolic component Auscultation: URSB enhanced S2, apex S4 Abdominal: (not found, would be secondary causes of hypertension) |
|
|
Methyldopa (prodrug), clonidine (direct)
|
= centrally acting sympatholytics
Mechanism: α2 agonists, inhibits sympathetic outflow from the brainstem Indications: HTN resistant to other treatments (decrease TPR and reduce CO) Side effects (significant so not really prescribed, adherence problems): dampen important sympathetic reflexes (positional hypotension), sedation (up to 50%), nausea, withdrawl syndrome (headaches, flushing, tachycardia, sweating, rebound hypertension |
|
|
Trimethaphan
|
= Peripherally acting sympatholytic
Mechanism: non-depolarizing inhibition of ganglionic nicotinic cholinergic receptors Indications: controlled hypotension (dissecting aneurysm, reduce surgical bleeding), dampen spinal autonomic reflexes (spinal cord injury) |
|
|
Prazosin (Minipress)
|
= selective vascular α1 adrenergic recptor antagonist
Mechanism: selectively blocks vasoconstrictor effect of sympathetic tone (allowing β2 relaxation to dominate) thereby causing apparent vasodilation and reducing TPR. Extent of vasodilation is limited by the degree of sympathetic activity causing vasoconstriction Indications: symptomatic prostate hypertrophy, monotherapy for mild/moderate HTN, polytherapy for HTN Side effects: improved lipid profiles, first-dose phenomenon of postural hypotension (impairs recovery from orthostatic hypotension) |
|
|
Propranolol (non-selective), Metoprolol (selective)
|
= β-adrenergic receptor antagonists (selective is for β1)
Mechanism: inhibits the inotropic and chronotropic effects of the SNS in the heart (β1), causing a reduction in HR and CO. Also inhibits SNS-mediated renin release in the kidney (lowering TPR) Indications: gold standard for HTN (use β1 selective for asthmatics) Side Effects: exercise intolerance (due to inhibited contractility), impotence |
|
|
Lisinopril (ACE), Losartan (AT1RB), Aliskiren (RI)
|
= Renin-Angiotensin system inhibitors.
Mechanisms: - ACEI's prevent ACE from converting Ang I to Ang II; while ATB's inhibit the AT1 receptor; Renin inhibitors bind renin (preventing Angiotens to Ang I conversion) - Net result of all is reduced volume (reduced Na+/H2O reabsorption) and TPR Indications: HTN Side effects: ACEI induce cough and angioedema (due to inability to breakdown bradykinin), also are teratogenic |
|
|
Diuretics
|
- Thiazide, amiloride (K+ sparing), Loop (Furosamide, Lasix)
Mechanism: act on the kidney to reduce volume of water retained and reduce TPR (major BP lowering mechanism) - Thiazide: block NaCl cotransporter in the distal tubule, not effective in renal failure - K+ sparing acts on Na transporters on distal collecting duct - loop diuretics: prevent the reabsorption of Na/K/(2)Cl in the ascending loop of henle - Acetazolamide: prevent bicarb reabsorption in proximal tubule, leading acidosis - Mannitol: osmotic diuretic, can't be resorbed by kidney directly reduced extracellular fluid Indications: mild HTN (thiazide) , adjuvant HTN therapy to (K+ sparing to prevent loss), refractory HTN (loop diuretics--reduce volume lots but not BP as much, cause hypokalemia so potentiate digoxin effects) Side effects: Thiazide causes increased glucose levels (contraindicated in pre/diabetes) |
|
|
Guidelines for therapy for Stage I & II HTN
|
Stage I:
- thiazide diuretic - alternate: ACE-I, ATB, β-B, Ca+ inhibitor. Consider patient preference and lifestyle (β are problematic for high activity) Stage II: - thiazide + ACE-I/ACB/β-B/Ca+ Inhibitor - evaulate patient compliance and tolerance for side effects |
|
|
Appropriate initial therapies for HTN in patients with comorbidities
|
Heart failure: thiazide, beta-blocker, ACE-I, ATB, aldosterone antagonist
Post MI: beta-blocker, ACE-I, aldosterone antagonist High cardiovascular disease risk: thiazide, beta-blocker, ACE-I, Ca+ blocker Diabetes: thiazide, beta-blocker, ACE-I , ATB, calcium channel blocker Chronic kidney disease: ACE inhibitor, angiotensin receptor blocker Recurrent stroke prevention: thiazide, ACE-I |
|
|
Cardiomyopathy
|
- any disease of the heart which result in mutated or non-functional elements of the cardiac sarcomere, specifically sliding filaments (actin/myosin) or accessory proteins.
May be genetic (hypertrophic, dilated, arrhythmogenic RV, restrictive, non-compaction) or acquired (CAD, HTN, valvular, viral, alcohol, chemo, peripartum, infiltrative) |
|
|
Familial dilated cardiomyopathy
|
- inherited condition which causes thining and dilation of the left ventricle (7-8cm vs 4-5) causing poor systolic function
- Genetics: usually autosomal dominant mutation of the sarcomere involving the myosin heavy chain (MYH6/7) and troponin (TNNT2). Can be screened for - cases are physiologically indistinguishable from acquired CM, must rely on FH for diagnosis Presentation: CHF or arrhythmia (Vtac esp concerning, fatal) due to structural abnormalities Exam: inferolaterally displaced apical impulse, S3 on auscultation (increased diastolic filling) Tx: standard Tx for CHF (β-B, ACE-I, diuretics), internal cardiac defibrillators (if arrhythmias are a risk) |
|
|
Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)
|
- autosomal dominant disease causing dilation/out-pouching and aneurysm of the RV wall (essentially diagnostic) due to fibro-fatty infiltrate.
- Often results in Vtach and sudden cardiac death (esp in athletes) - associated with mutations in desmoplakin, plakophilin-2, and desmoglein-2 (can be screened) - ECG: delayed V1 depolarizations (Epsilon waves, blip in early ST segment) from scar tissue in the RV |
|
|
Restrictive cardiomyopathy
|
- idiopathic or infiltrative (amyloid or sarcoid) genetic cardiomyopathy in which the ventricle is stiffened and less complaint (although normal thickness and contractility) leading to dystolic dysfunction over time.
Presentation: heart failure (dyspnea, volume overload, edema) - Difficult to differentiate from pericardial restriction. Patients will have normal size ventricles but huge, dilated atria on echo |
|
|
Left ventricular non-compaction
|
- developmental defect in which properly laid layers cardiac muscle do no compress, resulting in a ventricle with crypts and craters.
- complications: high risk for LV dilation and HF (due to poor muscle function), thromboembolic events (stasis in crypts), abnormal rhythms/SCD (if subjected to exercise) - genetics: not well understood, can be familial/sporadic, dominant/recessive/x-linked, sarcomeric/non-sarcomeric |
|
|
Hypertrophic cardiomyopathy
|
= genetic disease caused by mutation of one of several proteins of the cardiac sarcomere that results in LVH in the absence of another identifiable cause
- pathophys: systolic obstruction due to hypertrophied outflow tract and anterior motion of the mitral valve. Causing increased afterload and diastolic dysfunction - Epi: 1/500 adults, most common cause of SCD in young adults/athletes - Genetics: autosomal dominant mutation in one of 14 known genes encoding sarcomeric proteins: β-myosin heavy chain, myosin binding protein C, cardiac troponin T - Phenotype usually doesn't manifest until 2nd/3rd decade, and can be variable: usually asymmetric, most common is septal hypertrophy Histo: myocyte disarray with increased extracellular material - Brockenbrough's sign: increase in LV/Ao gradient during systole, increase in gradient/decrease in arterial pulse after premature beat |
|
|
Clinical features of HCM and treatment
|
- symptoms: dyspnea, angina (low perfusion due to thickened muscle), syncope, SCD, many are asymptomatic
- systolic outflow murmur: increased sound with stranding, valsalva, decreased with squatting, handgrip release, amyl nitrate - S4; due to atria contracting against stiff ventricle - Mitral regurg murmur - bisferiens/bifid pulse: due to mitral obstruction of the outflow (initially large volume dilates, then obstructed and needs more pressure) - flow meter: arcing systolic flow with notch in late ejection - EKG: high voltage QRS because of excess muscle, often abnormal/inverted T waves Therapy: - Rx: slow HR and negative inotropy: β-blockers (primary choice), Ca+ channel blockers (Verapamil, for non-obstructive patients), Disopyramide (negative inotrope) - surgical: myectomy (90% reduction in LV/Ao pressure gradient), alcohol septal ablation (risk of VT, pacemakers, coronary dissection) - other: internal cardiac defibrillator (indicated if at high risk) |
|
|
Risk factors for patients with HCM
|
- areas of ischemia/infarction due to hypertrophy causing scaring
- prior history of sustained VT or aborted SCD (necessitates pacemaker, ICD) Others: >2 indicates ICD - family history of SCD related to HCM - unexplained syncope - wall thickness >30mm - abnormal BP response to exercise (drops rather than increasing) - non-sustained VT on halter monitor |
|
|
Systolic dysfunction
|
= disease of myocardial contractility characterized by a reduction in the LVEF (<40%)
- normally maintained by hypertrophy or the Frank-Starling mechanism. Eventually these compensatory mechanism are overcome leading to phyisologic manifestations of HF: pulmonary congestion, secondary Rt heart failure - occurs in DCM can be seen in HCM (if LV dilation occurs) |
|
|
Dystolic dysfunction
|
- disease of abnormal ventricular filling characterized by elevated filling pressures (due to hypertrophy/decreased compliance)
- Characteristic of HCM and RCM, may occur in DCM - LVEF <40% |
|
|
Differential for cardiac masses
|
- Thrombus: most common, can be from MI, persistent AFib, hypercoagulable state, etc
- Non-bacterial thrombotic endocarditis: fibrin deposition on valves (do not destroy valves) - Bacterial endocarditis: ex: staph, if rt sided think IVD - Paradoxical embolus: in patients with patent foramen ovale - Inflammation - infection - sarcoidosis - foreign material (ex: bullet) - neoplastic growth (primary or metastatic) - tuberculoma (disseminated TB) |
|
|
Clinical manifestations of cardiac tumors
|
- Tumors typically present as impaired cardiac function. Ex: myxoma near valve causing valvular dysfunction.
- may have fever, elevated ESR (myxoma is associated with elevated peripheral inflammatory cytokines) - may manifest as distal emboli as they are often firable and easy to fragment, potentially leading to stroke or other ischemic event - Tumors can often be identified based location within the heart and age of the pateint - Primary cardiac sarcomas typically present w/ 5mo with non-specific symptoms preceeding acute exacerbation. Including: dyspnea, chest pain, tamponade, palpitations |
|
|
Cardiac myxoma
|
= most common cardiac tumor, benign
- majority present in the atria (esp the left) which may result in mitral valve dysfunction - multiple myxomas may be due to Carney Complex or Leopard Syndrome - typically manifest with: fever (elevated IL-6 and other cytokines), malaise, elevated ESR, valvular dysfunction. - These are friable tumors so at risk for embolism: stroke, GI infartion, distal extremity infartion Macro: penduculated or sessile and mobile causing changes in auscultation depending on position. Generally fleshy/fragile and pale gray to dark red, may be associated with calcifications Micro: myxiod matrix (gelatinous) with variable cellularity, may see mucin-secreting cells |
|
|
Angiosarcoma
|
- blood forming tumor, most common form of primary malignant cardiac tumor (but still very rare)
- usually affects people in middle age ~40 - poor prognosis: metastasis, not resectable, mean survival of 7mo to 2 years - often metastasize early to lungs, later vertebrae, liver, and brain - Presentation: tamponade (expectancy 3mo) - macro: violaceous, reddish brown lesion |
|
|
Cardiac rhabdomyoma
|
= most common form of pediatric cardiac tumor.
- benign, exclusive effect the ventricles and are usually multiple - clinical features depend on size and proximity to structures. Not uncommon for tumors to spontaneously remiss - often associated with tuberous sclerosis (autosomal dominany neurocutaneous syndrome, may cause cutaneous, renal, and brain lesions leading to seizures, mental retardation) Macro: circumscribed, unencapsulated white-yellow mass with waxy appearance in the wall of the ventricle. |
|
|
Carney Complex
|
= autosomal dominant condition associated with mutations in PRKAR1A gene resulting in cardiac and cutaneous myxomas
- disease is also associated with lentigenes (freckles), particularly in non sun-exposed areas. Family members will usually have them too - patients have characteristic blue nevi and melanotic schwannomas on skin - patients may also have endocrine disorders (Cushing's, pituitary adenoma), testicular tumors, mammary manifestations |
|
|
LEOPARD Syndrome
|
= genetic syndrome associated with cardiac masses and cutaneous lentigenes (freckles)
- cause EKG abnormalities and pulmonary stenosis - patients may also have ocular hypertelorism (extra space between eyes), gonadal hypoplasia, retarded growth, deafness |
|
|
Common malignant tumors that will metastasize to the heart
|
Cardiac metastasize usually result form fairly disseminated, high stage cancers.
- Usually involve the pericardium (rather than myocardium), except for melanoma and 50% sarcomas - often assymptomatic (discovered on autopsy/PET). May cause pleural effusion, conduction block, obstructions, ischemia, dyspnea, pericarditis - spread may be lymphatic (epithelial carcinoma, melanoma), hemodynamic (sarcoma, melanoma, renal cell carcinoma), direct extension (mediastinal: thymoma. esphageal), cavitary extension (via growth up IVC: renal cell carcinoma, hepatocellular carcinoma, adrenal and uterine tumors) - highest cardiac metatstatic rate is melanoma and malignant germ cell tumors |
|
|
Pleomorphic undifferentiated sarcoma
|
= second most common malignant cardiac neoplasm
- indifferentiated cell type (don't no origin) - primarily in the LA (<90%), especially the posterior wall - causes pulmonary congestion and mitral valve dysfunction - mean survival is 15mos. |
|
|
Layers of the aorta
|
Tunica intima: single layer of endothelial cells
Internal elastic lamina: composed of elastin and collagen, acts as a barrier to prevent movement of large proteins while contributing to structural integrity of the vessels Tunica media: smooth muscle layer, surrounded by another thin layer of elastin Adventitia: loose fibrous connective tissue, contributes to the load bearing of the vessel, also site of new blood vessel growth, possibly inflammation |
|
|
Changes in the aorta with age
|
= loss of compliance due to changes in the media and elastic wall from repetitive strain and relaxation. Elastic fibers become fatigued and fragment
- result is increasing systolic pressure and gradual declining diastolic, increasing pulse pressure - loss of compliance can result in isolated systolic HTN which is difficult to treat with medication due to the structural changes in the vessels (and may result in significant lowering of diastolic pressure) |
|
|
Abdominal aortic aneurysm
|
= most common site for aneurysm, involves all layers of the aorta and MMP (matrix metalloproteinas) activation and inflammatory reponse (Tcells and monocytes)
- distinct from thoracic aneurysms which have different pathology - Risk factors: male, old age, smoking (5x), caucasian, amputee, concurrent vascular problems (ED), family history of AAA or sudden death. NOT HTN Presentation: often asymptomatic, may have pain in legs, proximal gluteus, back Pathophys: changes in normal hemodynamic flow and atherosclerosis leading to turbulent flow and plaque deposit which activates chemokines and adhesion molecules (VCAM-1, AngIIR) causing inflammatory infiltrate, weakening all the layers of aorta leading to dilation/rupture - complications: rupture if over 5cm (greater than risk of surgical repair), mortality is 50% if ruptured |
|
|
Treatment for AAA
|
- open surgical graft: risk for damage to the spinal arteries which could lead to ischemia and paralysis
- endovascular stent-graft: does not alter progression of disease but bypasses circulation from the aneurysm (usually bifrucated to go down iliac ateries) - new approach: add polymer to adventitia to give structural support, doxycycline to inhibit MMP |
|
|
Diastolic heart failure
|
= abnormality of diastolic distensibility, relaxation, or filling of the LV + signs/symptoms of heart failure
Characteristics: normal LV filling volumes but higher pressures, normal LV chamber systolic function (EF>50%), concentric remodeling, abnormal diastolic filling/function Pathophys: reduced ventricular filling due to LVH/stenosis/reduced relaxation/restriction/reduced atrial contractility causing abnormal atrial distention and HF Diseases: LVF (HTN), HCM, RCM, ICM, transient M ischemia, MS, constricitive pericarditis, tamponade, obstructive myxoma Symptoms: vascular congestion (pulmonary), AFib |
|
|
Systolic heart failure
|
Characteristics: reduced systolic function (EF<50%, inadequate CO), progressive chamber dilation (Frank-starling), increased LV pressures (not as high as DHF) and eccentric remodeling (most patients also have diastolic dysfunction)
Pathophys: reduced systolic ejection due to obstruction/impaired contractility/chronic volume overload Diseases: MI, transient M ischemia, MR, AR, DCM, AS |
|
|
Clinical manifestations of heart failure
|
Symptoms are non-specific, have no other identifiable cause and tend to improve with diuresis
- dyspnea on exertion, paroxysmal nocturnal dyspnea, orthopnea, bilateral lower extremity edema, exertional fatigue, nocturnal cough Clinically cannot distinguish between systolic and diastolic heart failure: elevated JVP, pulmonary rales, S3 |
|
|
Definition of heart failure
|
= inability of the heart to pump blood forward to meet the metabolic and oxygenation requirements of the tissues while maintaining normal filling pressures backwards
|
|
|
Process of LV filling
|
Isovolumic relaxation: myocytes actively relax (ATP, ↓Ca2+ dependent) and elastic recoil (due to intrisinic compliance and geometry, against torque built in systole) restore ventricular size, lower pressure and promote active suction
Rapid filling: continued relaxation/recoil drop LVP<LAP opening MV and accelerating blood across (2/3 of filling) Diastasis: no driving gradient between LV/LA so slow/no filling, nearly absent in tachycardia Atrial systole/kick: contraction squeezing 25% of blood in, becomes more important if LV compliance is reduced |
|
|
Abnormalities associated/due to diastolic dysfunction
|
- slowed/delayed and incomplete myocardial relaxation
- decreased early diastolic suction/recoil - augmented LA pressure during early filling - shift of filling from early to late diastole (reliance on atrial kick) - Impaired rate and extent of LV filling (obstruction, reduced LV volume) - inability to significantly augment relaxation during exercise - increased diastolic LV, LA, and pulmonary venous pressures at rest or during exercise. |
|
|
Treatment of diastolic heart failure
|
- control of HTN
- control of ventricular rate (or rhythm), particularly in AFib - control of pulmonary congestion and peripheral edema: diuretics (but be careful of causing underfilling/hypotension in patients w/ small, stiff LV) - coronary revascularization in patient's who's CAD/ischema is impairing diastolic function (no evidence that it improves outcomes) - treat underlying disease (HTN, DM, etc) |
|
|
Compensatory mechanism in heart failure
|
Frank-starling mechanism:
- activated in HF that causes increased preload (DCM, MR, AR, hypervolumia, exercise). Increased volume stretches myoctyes (increasing sensitivity to Ca2+, optimizing myosin/actin overlap) increasing contractile force. Over time results in LVD and insufficient CO LaPlace law: - increased LVP or LVD stimulates wall hypertrophy/remodeling (ECM deposition) to maintain low tension (via laPlace's law) - Hypertrophy reduces compliance ↑diastolic pressures burdening LA and pulmonary vasculature Neurohormonal alterations: - sympathetic nervous system, renin-angiotensin-aldosterone system, vasopressin/ADH - respond to decreased BP by increasing salt/water retention, venous and arterial constriction, and HR which results in increased venous return ↑preload/afterload initially (helpful, Frank-starling), but increasing cardiac workload causes increased remodeling exacerbating HF |
|
|
Drug types/targets for treatment of heart failure
|
Therapy is to interfere with the neurohormonal compensation mechanism.
ACE-I/ARBs: block angiotensin II from increasing preload (via aldosterone and water/salt retention) and afterload (via SM vasoconstriction). **>10% Reduction in mortality risk (CHARM, VALIANT) Aldosterone receptor antagonists: inhibit salt/water retention in the kidney **30% mortality risk reduction (RALES) β1-Blockers: block sympathetic negative inotropy and chronotropy of the heart and renin activation in the kidney. **34% mortality reduction Diuretics (Thiazaide, Loop): block ion transporters in kidney tubules to reduce water retention. No effect of mortality Digitalis: increase cardiac contractility. No effect on mortality Vasodilators (Ca2+ channel blockers): prevent vasoconstriction and preload/afterload increase. No clear benefit Positive inotropes (epi, dobutamine): increase contractility in acute, severe HF |
|
|
Digitalis
|
Only drug that can increase contractility
Mechanism: 1. Na+/K+ATPase excretes Na+, 2. Digitalis glycosides blocks pump at K+ binding side causing intracellular Na+ accumulation, 3. Increased Na+ decreases gradient for Na/Ca exhanger, 4. Less Ca+ excreted, 5. extra Ca+ sequestered in sarcoplasmic reticulum, 6. more Ca+ released on muscle contraction, 7. Augmented cardiac contraction Indications: 2nd line drug: no survival improvement (worsen if discontinued) but treats signs/symptoms (CO, perfusion, tone, HR) Precautions: low therapeutic index, can promote arrhythmias, significant drug interactions, slow clearance (36-48hrs) |
|
|
4 classes of fluid in acute pericarditis
|
Serous (scant cells): most acute pericarditis starts this way, non-infectious inflammation, can be effusive/osmotic, rarely forms fibrous adhesions. From RA, SLE, CHF, thryroid dysfunction, nephrotic syndrome
Fibrinous: most common, due to chronic inflammation, forms adhesions/fused layers. Especially TB, also: AMI, post-infarction, uremia, radiation, RA ,SLE, trauma Purulent/suppurative: from bacterial infection (direct, hematogenous, lymphatic extension, or direct surgical introduction). Presents w/ fever, chills, spiking temp. Not common, rarely progresses to constrictive pericarditis Hemorrhagic: blood mixed with fibrinous or suppurative effusion. Most commonly from TB or direct neoplastic invasion. Alternatively: aortic dissection, tumors, trauma, uremia, cardiac surgery |
|
|
Etiologies of pericarditis
|
A: AIDS, Autoimmune disease (SLE, RA)
T: tumor, trauma, TB R: radiation, rheumatic fever I: idiopathic (usually means viral/post-viral), infection, infarction U: uremia M: medications (procainamide, hydralazine) |
|
|
Pericarditis following MI
|
Early onset:
- <1 week after MI, inflammation is an extension of the MI (spread to pericardium/epicardium). - more common in very large and/or trans-mural MI Dressler Syndrome: - >2wks after MI (can occur months later) - most likely autoimmune: antigens from dying cells activates delayed attack on pericardium and sometimes myocardium - simillar to post-pericardiotomy pericarditis Both are treated with high dose aspiring, be sure to distinguish from ischemia |
|
|
Physiologic mechanisms of acute pericarditis
|
Inflammation triggers:
1. local vasodilation: causing transudation of serous fluid 2. increased vascular permeability: protein spills into pericardial fluid 3. leukyocyte exudation: neutrophil and monocyte infiltration |
|
|
Key clinical findings in pericarditis
|
History: chest pain (pleuritic, worse when supine, better when sitting up/leaning forward)
Exam: soft heart sounds, friction rub (3 components: early systolic early/late diastolic, may blur together in fast HR, less sound w/ bigger effusions) Echo: effusion (%50) EKG: diffuse ST elevaqtion |
|
|
Pericardial effusion
|
"water-bottle heart," fluid visible on echo
Pathophys (non-pericarditis related): - ↑ capillary permiability: hypothyroidism - ↑ capillary hydrostatic pressure: CHF - ↓plasma oncotic pressure (↓albumin): nephrotic syndrome Symptoms: silent or symptomatic depends on volume and rate of fluid accumulation and the compliance of the pericardium Etiology: idiopathic (or post-viral) (39%), malignant (16%), radiation (7%), proven viral (6%), TB (4%) |
|
|
Cardiac tamponade
|
= a critical state of CV compromised due to ↑ pericardial fluid pressure compressing the heart continuously throughout the cardiac cycle and limiting ventricular filling
Pathophys: minimal stretch in the pericardium and ↑ pressure with ↑pericardial fluid causes increase and equalization of all the pressures in heart to the ↑interpericardial pressure (an pulm/venous system) during diastole. This limits ventricular filling and cardiac output Causes: acute ascending aortic dissection, any pericarditis w/ rapidly accumulating effusion (ATRIUM causes) Signs/symptoms: Pulseless, Beck's Triad: hypotension (paradoxical pulse), ↑JVP (deep X, flat Y), muffled heart sounds, tachycardia shock-like appearance (low-output), SOB, electrical alternans (on EKG), RA/RV collapse on Echo |
|
|
Paradoxical pulse
|
= Inspiratory drop in SBP (10mmHg) or marked decrease in pulse amplitude (karotkoff sounds go away during inspiration for > first 10mmHg)
Physiologic causes: inspiration drops intrathoracic pressure 1. ↓pulm vein pressure (effusion keeps ↑LV pressure) → ↓LV filling → ↓output and BP 2. ↑venous return (IVC/SVC) → ↑rt heart filling → shifts septum into the LV b/c effusion restricts expansion of free wall → ↓LV filling/output/BP Causes: tamponade, pericardial constriction, severe obesity (esp. supine), HF, asthma/COPD w/ air trapping, large pulmonary embolism |
|
|
Constrictive pericarditis
|
= thickening/fusion/calcification of the pericardium making it immovable with no measurable IPP. Results form chronic inflammation of pericarditis
Pathophys: impaired mid-late diastolic filling (no stretch to allow it), diastolic pressures in the heart and pulm system are elevated and equalized (limits ventricular filling/output), ventricular interdependence (ventricles push against each other, thoracic pressure not transmitted to heart so pulsus paradoxus) Presentation: cirrhotic-like appearance, ascites/edema, ↑HR, ↓BP/PP, ↑CVP/Kussmaul sign (↑JVP w/ inspiration), steep Y descent (dip and plateau), hard to find apex, KNOCK on ausculation (early/mid-diastole), calcification on CXR, dip/plateau on cath Other: effustive-constrictive pericarditis (rare): both high pressure effusion and stiff pericardium, see tamponade physiology until drained then constrictive |
|
|
Treatment for pericardial disease
|
Effusion: watch/wait, pericardiocentesis (drainage), pericardial window surgery
Pericarditis: NSAIDS, pain meds, indomethicin, steroids (often get rebound), cholcinine (good for chronic, microtubule inhibitor-stops macrophages) Tamponade: pericardiocentesis, pericardial window Constrictive pericarditis: pericardial stripping surgery |
|
|
Virchow's triad
|
Factors that increase risk of thrombosis/DVT
1. Venous stasis: immobilization (paralysis, spinal cord injury, stroke, long trips), age >40, varicose veins, severe COPD, anesthesia, MI/CHF, obsesity 2. endothelial damage: surgery, inflammatory state, prior DVT, central line, trauma, major venous surgery, smoking 3. hypercoagulable state: genetics (AT111, protein C/S, factor V leiden, antiphospholipid AB, homocystinuria), cancer, estrogen, IBD, nephrotic syndrome, sepsis, blood transfusion, inflammatory disease, heparin-induced thrombocytopenia |
|
|
Clinical prediction of DVT
|
Stratify with Wells Criteria: active cancer, immobilization, bedridden from surgery >3d, local tenderness along veins, unilateral swelling/pitting, collateral superficial veins, Alternative Dx more likely (-2)
For high/moderate risk get D-dimer (if normal no further testing) Abnormal D-dimer: ultrasonography, repeat in 1 week if normal |
|
|
Risk factors for DVT and PE
|
DVT: Virchow's triad
PE: - recent surgery, especially if new dyspnea/hypoxia/tachycardia - known proximal DVT (above knee) - known RV failure (high risk for new DVT/PE) - known malignancy, especially breast/pancreas/ovary/lung - known hypercoagulable disorder - post-partum women or people on estrogens, especially if new dyspnea |
|
|
Progression of DVT
|
1. Underlying risk factor leads to platelet activation and thrombosis
2. with small clot, patient is asymptomatic and limb is supplied by collaterals 3. as clot grows/becomes occclusive it obstructs flow asymetrically and patient becomes symptomatic: unilateral swelling, pain, ulceration (if prolonged), postive Homan's sign 4. may break off and embolize to lungs |
|
|
Clincal presentation of PE
|
symptoms: dyspnea, pleuritic chest pain, cough. apprehension, syncope
signs: often asymptomatic, rales or crackles, tachycardia, diaphoresis, hemoptysis |
|
|
Bedside findings of PAH
|
General: clubbing
Arterial: hypokinetic carotid pulse Venous: giant A or CV/systolic wave (due to TR) Precordial: absent apex (low CO), parastrenal lift, palpable pulmonary artery pulse at ULSE Auscultation: - LUSB: loud S2 (from P2, palpable when severe), delayed P2 w/ RV failure, ejection sound, PR murmur (Graham Steell) - LLSB: S4 (rt sided), TR murmur If RF: hepatomegaly/pulsatile liver, elevated JVP, peripheral edema, ascites, pleural effusion, lower extremity edema |
|
|
Diagnostic tests for PAH
|
- EKG: tall R wave in V1-4, ST segment abnormalities, inverted VI due to RA dilation, tall lead II P wave to RA abnormality
CXR: prominent pulmonary vessels (diminished in periphery), RV enlargement (less retrosternal space), RA elargement (prominent rt border) Echo: dilated RV with flatten septum (into LV) Catheterization: measure RAP, RVP, PCWP |
|
|
Symptoms of PAH
|
- dyspnea and fatigue: due to ↓CO, hypoxemia, and ↑work of breathing
- angina: ↑O2 demand of RV and inadequate delivery (↓CO, hypoxia), LV may also become ischemic when severe - Syncope: ↓CO so ↓BP during normal vasodilation (exercise, heat), also from arrhythmias (AFib: dilated RA, VT: dilated/ischemic RV) - Edema: RV failure causes peripheral accumilation, and due to deep venous insufficiency |
|
|
WHO classification of PAH
|
Group 1: Primary, PAH
- mPAP ≥25, PCWP ≤15, no CHF, obstructive/restrictive lung disease, thromboembolic disease Types: idiopathic, heritable, drug/toxin induced, associated w/: CTD, HIV, congenital HD, peristent newborn PAH, schistosomiasis, hemolytic anemia, portopulmonary disease Group 2: LH disease - mPAP ≥25, PCWP >15 - heart diease: valvular, LV systolic or diastolic dysfunction Group 3: lung disease - COPD, interstitial, apnea, altitude, developmental, alveolar hypoventilation, other obstructive/restrictive Group 4: chronic thromboembolic - uncommon, but surgically treatable (remove clot/damage). Mechanism is pulmonary arterial obstruction Group 5: PH w/ unclear or multifactorial causes - chronic myeloproliferative disease, splenectomy, sarcoidosis, pulmonary langerhans cell histiocytosis, LAM, glycogen storage disease, hyper/hypothyroidism, end-stage renal |
|
|
Molecular mediators involved in pathogenesis of PAH
|
Vasoconstrictors (increased activity: enodothelin, thromboxane A2, inflammation, oxidation stress, etc
Vasodilators (reduced activity): prostacyclin (and synthase), NO (and synthase), fibrinolysis |
|
|
Peripheral vascular disease: definition and classification
|
= disorders of the circulatory system to the extremities, viscera, and head
1. structural changes in the vessel wall due to degenerative conditions, infection or inflammation leading to dilation, aneurysm, dissection or rupture 2. narrowing of the vascular lumen caused by atherosclerosis, thrombosis, or inflammation 3. spasm of the vascular smooth muscle. |
|
|
Presentation of atherosclerotic PAD
|
PMH: smoking
Symptoms: - often asymptomatic initially - claudication (from ischemia): pain, tenderness, soreness, aches, weakness, tiredness, numbness - classically in the calves (disease one level above), exercise induced progressing to pain at rest, reproducible PE: - loss of distal pulses, bruits over stenotic arteries, ankle-brachial index (divide SBP a/b, >.9 normal, .4-.7 mod obstruction, predicts ischemic events) - in severe cases: muscle atrophy, pallor, cyanosis, hair loss, ulceration, distal necrosis, and amputation |
|
|
Treatment for PAD
|
- Goals: improve functional status/QoL, preserve limb, prevent severe complications of atherosclerosis (MI/stroke)
- Risk factor modification: smoking, DM, cholesterol, BP, exercise, wound care - Medical management: treat contributing conditions (DM, dyslipidemia, HTN), prevent complications: ACE-I, antiplatelet therapy - Surgical intervention: endarterectomy, bypass, thombectomy, ligation - Minimally invasive techniques: angioplasty/balloon, stent, thrombectomy/lysis, embolization/ligation - Follow-up care: risk factor modification, medical management, status assessment |
|
|
Risk stratification for carotid revascularization
|
Symptomatic patients: patients with stroke or TIA
- NASCET trial: prevents 1 stroke for 6 patients (70-99% stenosis) in 2 yrs. Less occluded patients (50-69%) 1/22 at 5 years - co-morbidities adversely affects ourcomes, multiple contributes to higher post-op mortalitiy Asymptomatic: - ACST: patients w/ high stenosis (>60%) could have benefit (made in 1989 before current medical management) - maximal benefit soon after stroke (<1mo), unless massive stroke to prevent reperfusion injury/hemorrhage - can be operated on if their risk of stroke > greater than risk of procedure (post-op 2%) Endarterectomy vs. Stent (prior surgery/radiation, severe cardiopulm disease): - general vs. local anesthesia, shunt vs. none (intact circle of willis?, patch options - CREST: no difference in primary outcomes (mortality, stroke, MI), but endarterectomy has higher MI rate (maybe just troponin bumps?), stenting higher stroke risk (more clinically debilitating) |
|
|
Risk factors for atherosclerotic disease
|
- tobacco use, elevated cholesterol, age (increases w/ 10years), high BP, diabetes, obesity, high fibrinogen levels, male gender, physical inactivity
- alcohol seems protective? |
|
|
Systematic review
|
= a literature review focused on a research question that tries to identify, appraise, select and synthesize all the high quality research relevant to that question. Aims to provide an exhaustive summary of literature relevant to a research question.
|
|
|
Review article
|
= opinion piece of the evidence available about a topic. The author is not obligated to include all research about a topic, rather may cherry pick sources, therefore introducing bias into the analysis
|
|
|
Meta-analysis
|
= a statistical analysis of a collection of analytic results for the purpose of integrating findings. Combining studies give a lot of power to the study (by pooling many samples), and drops type II error (rejecting alternate hypothesis when it is true)
Methodology: - systematic literature review: list databases and citation indexes used, an attempt should be made to look at unpublished studies - evaluate selected against inclusion/exclusion criteria (should be made explicit in methods section) - assign an assessment of quality to each paper using an objective rating system (forest or funnel plot, multiple evaluators) |
|
|
Forest plot
|
= graphical display designed to illustrate the relative strengths of treatment effects of multiple studies addressing the same question (graphical representation of meta-analysis results)
- Odds ratio for the results of each study is plotted on the bottom. Squares represent the weight of the study in the meta analysis, and the horizontal lines the diffidence interval |
|
|
Funnel plot
|
= scatter plot of treatment effect against a measure of study size.
- designed to check for publication bias or heterogeneity in systematic reviews and meta-analyses. - assumes that the largest studies will be near the average while smaller studies will be spread on either side of average. A symmetric funnel shape should arise from a good data set (indicates publication bias is unlikely) - asymmetric funnel indicates publication bias or a systematic difference between small and large studies ("small study effects") or from an inappropriate effect measure. |
|
|
Publication bias
|
the tendency of researchers, editors, and pharmaceutical companies to handle the reporting of experimental results that are positive (i.e. showing a significant finding) differently from results that are negative (i.e. supporting the null hypothesis) or inconclusive, leading to bias in the overall published literature
- Such bias occurs despite the fact that studies with significant results do not appear to be superior to studies with a null result with respect to quality of design. - The most common reason for non-publication is an investigator's declining to submit results for publication (because of the investigator's loss of interest in the topic, the investigator's anticipation that others will not be interested in null results, etc.), underlining researchers' role in publication bias phenomena. |
|
|
Study weighting
|
- there are other differences between the studies that need to be allowed for, but weighting allows a more powerful estimate the true effect size—as opposed to a less precise effect size derived in a single study under a given single set of assumptions and conditions
- A meta-analysis typically combines the results of several studies that address a set of related research hypotheses by identification of a common measure of effect size, of which a weighted average might be the output of a meta-analysis. The weighting might be related to sample sizes within the individual studies |
|
|
Quality weighting
|
- not all studies are of equal quality so weighting is necessary to not disrupt the overall analysis with poorly collected data
- Be wary of studies that only report results in terms of quality scores to weigh study effect, as quality scoring can introduce significant bias and should be sued more for study exlusion than for adjustment of the results of any individual study. When quality scores are included, they should be clarified in terms of sample size studied and confidence interval breadth - Qualitative variables that must be weighted included whether the trials reported were blinded, funded by an impartial agency, whether they involved random subject selection and assignment, and whether the study had a low drop-out rate. |
|
|
Kappa Statistics
|
= a statistic that takes into account the fact that observers in a study will sometimes agree or disagree simply by chance
- A kappa of 1 indicates perfect agreement, whereas a kappa of 0 indicates agreement equivalent to chance. > 0.8 means reviewers agree and K <0.4 indicates poor agreement. At least 2 coauthors should assess every study and rate their degree of agreement on the quality of that study |
|
|
Tests of heterogeneity
|
A kappa of 1 indicates perfect agreement, whereas a kappa of 0 indicates agreement equivalent to chance. > 0.8 means reviewers agree and K <0.4 indicates poor agreement. At least 2 coauthors should assess every study and rate their degree of agreement on the quality of that study
|
|
|
Non-infectious endocarditis
|
- rheumatic fever: non-bacterial thrombotic endocarditis: verrucae from on the endcardium which facilitate infections (leading to infectious endocarditis from increased surface area for bacterial adherence)
- SLE: Libman-Sacks endocarditis: verrucous vegetations along the endocardium including the valves - Cancer: marantic endocarditis: sterile, non-destructive vegetations (small thrombi) Both NBTE and marantic endocarditis present with sterile, non-destructive vegetations (small thrombi on endocardium), leaving the valves functionally normal. Can be associated with hypercoagulable state, SLE, and mucing producing adenocardinoma |
|
|
Infective endocarditis
|
Subacute:
- idolent (weeks-months) infection, usually S. viridans (non-virulent) (or S. Bovis, HACEK) of valves with prior damage. - Symptoms: indisidous, low-grade fever, malaise, anorexia, night sweats (may be mistaken as another illness/flu). Valve destruction is mild to moderate Acute: - Fulminant infection (days), usually S. aureus (virulent) of usually normal valves - Symptoms: abrupt fever, chills, very ill, severe valve destruction Pathogenesis: 1. NBTE formation: turbulent flow leading to endothelial damage and platelet-fibrin deposits. 2. transient bacteremia (will have positive blood cultures): trauma to mucosa releases microbes. 3. Bacterial adherence: via adhesins/agglutinins, localized to high flow, low pressure side. 4. Vegetations formed: composed of fibrin/platelets/red cells/bacteria. 5. Bacterial proliferate and cause valve damage (valvular damage can be heard or visualized on echo usually) Special situations: 1. IVDA: acute S. aureus infection of TV +/- septic emboli 2. Prosthetic heart valve: highest risk <6mo (<12mo S. epi, >12mo S. aureus), 3-5% of valves (mechanical = prosthetic, AV = MV), perivalvular infection commmon 3. Culture negative: 2-7%, C. burnetii or Bartonella sp, etiology (previous abx, inadequate microbio techniques, fastidous/fast growing organisms), 50% survival if febrile >1wk 4. Elderly: increased incidence and mortality, insidious onset, risks (hospitalized, invasive procedures), entry (GI, GU) 5. Children: less common/less mortality except premies (S. aureaus + ToF + P. Atresia), improved survival |
|
|
Clinical manifestations of endocarditis
|
F: fever
R: Roth spots/petechiae (small interocular/congunctival hemorrhage) O: Osler nodes (Painful, violaceous nodules on fingerpads/toes, immune complex deposits) M: murmur/regurg (new or changing) J: Janeway lesions (hemorrhagic, painless, flat plaques on palms and soles, septic emboli) A: abcess (myocardial), aneurysm (mycotic), anemia N: nailbed ("splinter") hemorrhages and neurologic problems E: emboli (usually septic), elevated ESR |
|
|
Duke criteria for diagnosis of infective endocarditis and treatment
|
Major criteria
1. Positive blood culture (2 separate cultures) 2. evidence of endocardial involvement (positive echo or new regurgitation) Minor criteria: predisposing cardiac condition (cyanotic HD) or IVDA, fever >38.0, vascular phenomena (septic emboli, mycotic aneurysm, intracranial hemorrhage, congunctival hemorrhage, janeway lesions), immunologic phenomena (glomerulonephritis, olser nodes, roth spots, RF+), positive blood cultures not meeting major criteria Tx: - Abx +/- valve replacement (refractory HF, >1 embolic episode, uncontrolled infection, lack of effective antibiotics, most infected prosthetic valves) |
|
|
Complications of infective endocarditis
|
Valve destruction:
- regurgitant lesions leading to heart failure - aneursym, perforation or abcess Local extension of in the infection within the heart: - progressive valvular damage, abscess formation, erosion into the conduction system (arrhythmias and contractility problems) Peripheral emboli (or immune complex deposition): - infarct, aneurysm, or vasculitis of distal tissue (usually brain, kidney, spleen) |
|
|
Mechanisms of arrhythmia
|
1. Abnormal automaticity (atrial or ventricular): ectopic foci with chaotic behavior can cause additional beats or dominate all together
2. Triggered activity (atrial or ventricular): disturbances of repolarization trigger regular response activity. - early depolarization: during phase 3, associated with factors increasing AP duration (long QT syndrome) - Delayed depolarization: during phase 4, associated with high intracellular Ca2+ (digitalis, pheochromocytoma) 3. Re-entry disturbance: a region of cardiac tissue with unidirectional block allowing retrograde activity causing additional activity. Caused by structural defect or previous mycardial infarct |
|
|
Classes of Antiarrhythmetics
|
Class I: sodium channel blockers (quinidine, lidocaine, propafenone)
- slow the rise of action potential slowing conduction, reduce HR (raise AP threshold) - pro-arrhythmic, take w/ Beta blocker to slow HR Class II: Beta-receptor blockers (propranolol, metoprolol, atenolol) - inhibit sympathetic stimulation of the SA node, slow sinus rhythm Class III: Potassium channel blockers (amiodarone, sodolol) - prolong QT interval, increasing refractoriness - can be pro-arrhythmic (torsades, VT, polymorphic QRS, PVC) - lots of side effects Class IV: Calcium channel blockers (verapamil, diltiazem) - block channels mediating SA/AV node depolarization, slowing sinus rhythm and AV-nodal conduction velocity Misc: Adenosine - acts on G-protein coupled receptors in the AV node causing K+ channel depolarizatoin (sinus bradycardia), slowing AV conduction, and inhibiting re-entry |
|
|
Class I Antiarrhythmics
|
= block Na+ channel, preventing flux and slowing AP upstroke/conduction velocity. Cause negative inotropy and increase AP threshold
- preferentially bind active channels (more active tissue) - These can be pro-arrhythmic so use w/ Beta-blocker to slow HR and prevent Afib-Vfib conversion 1A: quinidine: - indicated for AFib/Aflutter, not for hypotension (alpha block) or long QT. - Side effects: diarrhea, lupus, cinchonism, torsade de pointes IB: lidocane - indicated for ventricular arrhythmias (post-MI), not for Stoke-Adams WPW - side effects: CNS disturbance IC: propafenone - indicated for SVT, not for MI or long QT - side effects: worsens life-threatening arrhythmias |
|
|
Class III Antiarrhythmics
|
= block K+ channels, prolonging QT and increasing refractoriness
- these can be pro-arrhythmic but generally less than class I - requires high dosing (non-selective, huge volume of distribution), long half-life Amiodarone: - indicated for SVTs (Afib/flutter), ventricular arrhythmias, adj therapy w/ ICD. Nor for iodine sensitivity, thyroid or pulm disease, Long QT - side effects: pulm fibrosis, thyroid dysfunction, peripheral neuropathy, liver toxicity, ocular deposits, torsades de pointes, ashen/blue skin |
|
|
Class III Antiarrhythmics
|
= beta blockers (propranolol, metoprolol, atenolol). Inhibit sympathetic stimulation of the SA node
- Indicated for: SVTs (sinus tachycardias and others). Not for: severe bradycardia (CHF) - Side effects: bradycardia, hypotension |
|
|
Class IV Antiarrhythmics
|
= Ca+ channel blockers (not dihydropyridines/nifedipine) block channels in SA and AV nodes slowing signal propigation, prolongin PR interval
Indicated for: SVTs (controls ventricular rate). Nor for severe hypotension, WPW Side effects: hypotension, negative inotropy |
|
|
Adenosine
|
= antiarrhythmic, acts on G-protein coupled receptors in the AV node causing K+ channel depolarizatoin (sinus bradycardia), slowing AV conduction, and inhibiting re-entry
Indicated for: SVTs, diagnostic sources of SVT. Not for severe AV block, hypotension, bronchospasm - Side effects: relatively minor |
|
|
Treatment options for different types of arrhythmias
|
Afib/flutter:
- acute: diltiazem, beta-blocker (control HR), cardioversion - longterm: class IA, IC, III, surgical ablation Other SVTs: - acute: vagal stimuli, adenosine, verapamil, diltiazem - longterm: cardioversion or atrial pacing, class IA, IC, II, IV Sustained VT: - acute: lidocaine - long term: ICD, amiodarone, beta-blocker VF: - defibrillation - ICD, amiodarone, beta-blocker |
|
|
Progression of MI on ECG
|

|
|
|
Major side effects of CV drugs
|
Swollen tongue / cough- ACE I
Pericarditis (pleuritis) - hydralazine, procainamide HypoT4 / skin color- amiodarone Leg edema- nifedipine Loss of appetite / ECG abnormalities - digitalis |