Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
175 Cards in this Set
- Front
- Back
|
susceptibility of a fetus to teratogens depends on what three factors?
|
1. genotype
2. # of interactions with teratogen 3. developmental stage of fetus |
|
|
Describe the susceptibility of teratogens during the following fetal times:
1. 0-14 days (preimplantation and implantation) 2. 15-60 days (early and late organogenesis) 3. 60-280 days (fetal period) |
1. resistant time for embryo: all or nothing response
2. MOST SUSCEPTIBLE TIME for fetus 3. not as susceptible |
|
|
define:
1. dysraphic anomalies 2. atresia |
1. defects caused by the failure of apposed structures to fuse
2. defects caused by incomplete formation of a lumen. |
|
|
what are two possible results of defects in either cell proliferation or induction?
|
1. death
2. agenesis |
|
|
a defect in cell differentiation would result in? (2)
|
Agenesis
OR Hamartomata (tumor growth) |
|
|
what are three possible results of a defect in migration?
|
1. non-fusion
2. fusion 3. misplacement |
|
|
during embryonic development, a defect in cell death would result in?
|
persisting transitory structures
|
|
|
a theory postulating how FAS develops involves inhibition of retinol dehydrogenase, thereby resulting in a lack of product.
What is the product and why is this product important? |
retinoic acid
important in homeostasis and normal fetal tissue development. |
|
|
what is a physical sign seen in newborns that almost always indicates intracranial pathology?
|
fisting
|
|
|
What does TORCH stand for?
|
Toxiplasmosis
Others Rubella Cytomegalovirus Herpes simplex |
|
|
what is the diagnostic tool for CMV in an infant?
|
intranuclear inclusion bodies in urine sediment.
|
|
|
what are some common effects seen in a newborn that is infected with a TORCH agent?
|
Microcephaly, focal cerebral calcifications
Cataracts, chorioretinitis hepatosplenomagaly heart disease purpura and petechiae |
|
|
a pregnant woman that has ingested Thalidomide has (in general) a ___% risk of having a child with birth defects?
|
20%
|
|
|
What is the timeperiod in the pregnancy that thalidomide is a teratogen?
|
21-33 days post conception
|
|
|
what are the hallmark clinical manifestations of a thalidomide baby?
|
microtia or anotia
phocomelia or amelia (flipper or no arms) cardiac defects |
|
|
describe the mental status of a child with:
1. FAS 2. CMV 3. Thalidomide exposure |
1. MR, learning disabilities, ADHD
2. MR 3. Normal intelligence |
|
|
4p- syndrome is also known as?
|
Wolff-Hirschkoff syndrome
|
|
|
Hallmark phenotype of a child with 4p- syndrome?
|
growth retardation
"Grecian Helmet" facies hypertelorism, cleft lip or palate |
|
|
Which disorder is characterized by a clenched fist with overlapping digits (2nd over 3rd, 5th over 4th)?
|
Trisomy 18
(nondisjunction) |
|
|
what is another hallmark phenotype of a child with trisomy 18?
|
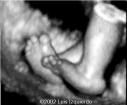
rocker-bottom feet
(also short sternum, cardiac defects) |
|
|
which disorder is characterized by midline facial defects?
|
trisomy 13
|
|
|
what is another hallmark phenotype of a child with trisomy 13?
|
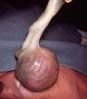
omphalocele
(hyperconvex nails, microcephaly) |
|
|
compare the mortality rates of trisomy 13 and 18.
|
trisomy 13 - 50% mortality first month
trisomy 18 - 95% mortality first year most of these fetuses (66-75%) are lost during pregnancy |
|
|
what is the genotype of an individual with Klinefelter Syndrome?
|
47,XXY
|
|
|
Describe the morphology and function of the testes in an individual with Klinefelters
|
Seminiferous tubules: atrophy, hyalinization, fibrosis
Germ Cells and Sertoli Cells: absent Leydig cells: increased in number, function impaired |
|
|
what would the following hormone levels be in an individual with klinefelters?
1. Testosterone 2. Estradiol 3. LH, FSH |
1. Low
2. High 3. High |
|
|
hallmarks in the phenotype of a Klinefelter individual?
|
gynecomastia
tall, eunochoidal phenotypic male genitalia female escucheon |
|
|
mental status of a klinefelter individual?
|
reduced IQ
behavioral and personality problems |
|
|
what is the genotype of an individual with Turner Syndrome?
|
45,X
(3/4 of X's maternal origin) |
|
|
clinical features of Turner?
|
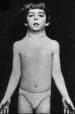
short stature
web neck coarctation of aorta, bicuspid aortic valve rudimentary ovaries multiple nevi |
|
|
mental status of an individual with Turner's?
|
normal IQ
some learning disablities (esp. math, visual-spacial) |
|
|
Wardenburg's syndrome is caused by a failure of normal neural crest cells to migrate and differentiate. What are three cell lines derived from neural crest cells that are affected?
|
1. Melanocytes
2. Colonic Ganglion Cells 3. Hair cells in Organ of Corti |
|
|
what is the inheritance of Wardenburg Type I syndrome?
|
autosomal dominant
|
|
|
Clinical features of Wardenburg Type I?
|
white streak in hair
unilateral/bilateral deafness hypopigmented ocular fundus Hirschprung's disease (colonic aganglionosis) |
|
|
Dandy-Walker malformation is a type of?
|
hydrocephalus
|
|
|
the genetic inheritance of hydrocephalus is varied:
1. genetic defect specifically resulting in atresia of the foramina of Luschka and Magendie? 2. what about Congenital Sylvius aqueductal stenosis? |
1. autosomal recessive
2. X-linked recessive |
|
|
Clinical hallmarks of hydrocephalus?
|
Large tense fontanelle, ventricular enlargement
"sunsetting" of pupils" sixth nerve palsy: limitation of upward gaze hyperactive deep tendon reflexes |
|
|
what is the theorized pathogenesis of hydrancephaly?
|
early fetal obstruction of carotid vessels: results in no cerebral tissue. Brainstem still present b/c basilar arteries are not blocked.
|
|
|
clinical manifestations of hydrancephaly?
|
transillumination
enlarged head, large fontanelle preservation of MORO reflex, rooting and sucking |
|
|
treatment for hydrancephaly?
|
shunt
(still observe progressive deterioration, most deaths due to pneumonia) |
|
|
what type of genetic disorder is myotonic dystrophy (MD)
|
autosomal dominant
trinucleotide repeat expansion |
|
|
which enzyme is defective in MD?
|
myotonia protien kinase
|
|
|
how does "characteristic myotonia" manifest in an EMG?
|
"dive bomber" sound
|
|
|
Clinical hallmarks of myotonic dystrophy?
|
myotonia w/sustained grip (handshake that is slow to relax)
facial weakness, inability to bury eyelashes ptosis, cataracts |
|
|
what is the inheritance of congenital myotonic dystrophy?
|
autosomal dominant
|
|
|
clinical manifestations of congenital myotonic dystrophy?
|
severe hypotonia
facial weakness floppiness poor feeding respiratory insufficiency |
|
|
what type of mutation is observed in spinal muscular atrophy and what structure is affected by this mutation?
|
deletion mutation
Reduction in survival motor neuron (SMN) protien |
|
|
what would you expect the motor nerve conduction to be in SMA?
What would you expect the EMG results to be? |
normal
(EMG shows denervation) |
|
|
clinical hallmarks of SMA I?
|
hypotonia
no reflexes respiratory problems, diaphragmatic breathing cannot sit alone |
|
|
what is a major clinical difference between SMA I and SMA II?
|
SMA I - earlier onset, child cannot sit
SMA II - later onset, child can sit |
|
|
compare the age of death in SMA I, II and III.
|
SMA I - less than 2 yrs
SMA II- greater than 2 yrs SMA III- adult |
|
|
Compare the muscle biopsies of SMA I, II and III.
|
SMA I and II similar, see clusters of large fibers
SMA III - more normal |
|
|
clinical manifestations of SMA III?
|
can walk with braces
proximal weakness, utilize Gower maneuver weakness |
|
|
what are the distinguishing features between spinal muscular atrophy (SMA) and muscular dystrophy?
|
muscular dystrophy patients have large calves;
SMA pts. do not both utilize Gower maneuver |
|
|
what is the disorder?
- impaired rod/cone function - can be autosomal dominant, recessive or X-linked recessive |
retinitis pigmentosa
|
|
|
clinical manifestations of retinitis pigmentosa?
|
tunnel vision
decreased dim light vision diffuse retinal depigmentation affected color vision |
|
|
what is the avg. time of onset for retinitis pigmentosa?
|
1st-2nd decade
(result is severe vision loss 4th-7th decade) |
|
|
Appearance of Lisch nodules is characteristic of?
|
Neurofibromatosis I
|
|
|
what is the genetic defect in neurofibromatosis I?
|
defect in NFI gene
|
|
|
pathophysiology of neurofibromatosis?
|
neurofibromin protien normally inactivates Ras: when neurofibromin is defective Ras activity is uncontrolled and neurofibromas form
|
|
|
what other clinical manifestations are present in neurofibromatosis I?
|
cafe-au-lait spots
skeletal lesions (lisch nodules, neurofibromas) |
|
|
what is a plexiform neurofibroma (vs. a regular neurofibroma)?
|
plexiform neurofibromas are cutaneous and raised, neurofibromas are not
|
|
|
which disease is known for its characteristic "candle drippings"? (subependymal nodules)
where are these found? |
Tuberous Sclerosis Complex (TSC)
candle drippings are found on ventricular walls |
|
|
which genes are mutated in TSC?
what do these genes encode? |
TSC 1 OR TSC 2
both encode tumor supressor proteins |
|
|
what is the inheritance of TSC?
|
autosomal dominant
|
|
|
pathophysiology of TSC?
|
disordered or arrested migration of neuroectoderm
|
|
|
Clinical manifestations of TSC?
|
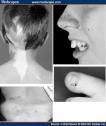
facial angiofibromas "adenoma sebaceum"
cardiac rhabdomyomas kidney angiomyolipomas ash leaf spots |
|
|
which disorder is characterized by a port wine stain along the trigeminal nerve?
|
Sturge Weber
|
|
|
what is the primary problem in Sturge Weber disorder?
|
vascular plexus abnormality
|
|
|
Other clinical manifestations of Sturge-Weber?
|
intracranial hemangioma, calcification
MR |
|
|
which disorder is characterized by abnormal vesicles in the epidermal layer secondary to recurrent minor trauma?
|
Epidermolysis Bullosa Simplex
|
|
|
inheritance pattern of EB?
abnormal protien? |
autosomal dominant
abnormal keratin |
|
|
what is the progression of EB immediately following a minor trauma?
|
cytoplasmic vacuoles
coalescence rupture of plasma membrane leading to cell lysis intraepidermal vesicles healing w/o atrophy or scarring |
|
|
ichthyosis vulgaris is a disorder resulting in defects of which two molecules?
|
profilaggrin
filaggrin |
|
|
Which disorder am I?
rocker bottom feet |
trisomy 18
|
|
|
Which disorder am I?
purpura and petechia |
TORCH
(specifically CMV) |
|
|
Which disorder am I?
Candle drippings (subependymal nodules) |
Tuberous Sclerosis Complex (TSC)
|
|
|
Which disorder am I?
port wine stain? |
Struge Weber
|
|
|
Which disorder am I?
Dandy-Walker malformation |
Hydrocephalus
|
|
|
Which disorder am I?
Grecian Helmet facies |
4p-
(Wolff-Hirschkoff) |
|
|
Which disorder am I?
cafe-au-lait spots |
neurofibromatosis
|
|
|
Which disorder am I?
tunnel vision, decreased light vision, abnormal ocular pigmentation |
retinitis pigmentosa
|
|
|
Which disorder am I?
Gower maneuver |
spinal muscular atrophy III
OR muscular dystrophy |
|
|
Which disorder am I?
white forelock |
Waardenburg Type I
|
|
|
Which disorder am I?
47 XXY |
Klinefelter Syndrome
|
|
|
Which disorder am I?
midline facial defects |
trisomy 13
|
|
|
Which disorder am I?
"basically anencephaly with the skull closed" |
hydranencephaly
|
|
|
Difference between malformation and deformation?
|
malformation - caused by abnormal development
deformation - caused by mechanical forces |
|
|
What is the name of the disease that is due to abnormal zinc absorption?
|
Acrodermatitis Enteropathica
|
|
|
what is the precise reason why there is abnormal zinc absorption in an individual with acrodermatitis enteropathica?
|
there is an absence of low molecular weight zinc binding factor
|
|
|
In an individual with acrodermatitis enteropathica, what would you expect the serum zinc levels to be?
|
low
|
|
|
what are some hallmark symptoms seen in acrodermatitis enteropathica?
|
- vesiculopustular erruptions (mouth, anus, genitals, distant extremities)
- alopecia - severe diarrhea - abnormal humoral/cellular immunity - lethargy |
|
|
is there a treatment for acrodermatitis enteropathica?
|
administer oral Zinc
|
|
|
which disorder is a result of a missense mutation in the fibrillin-1 gene?
|
Marfan syndrome
|
|
|
what are some skeletal manifestations of Marfan syndrome?
|
- tall stature
- arachnodactyly - joint hypermobility - pectus excavatum or carinatum - walker-murdoch wrist sign - steinburg thumb sign |
|
|
what is the occular hallmark that suggests Marfan syndrome?
|
upward lens subluxation
|
|
|
describe the cardiac mainifestations of marfan syndrome
|
aortic dilitation, regurgitation, often results in aneurism of aortic root
|
|
|
which disorder is characterized by deficient or defective collagen?
|
Ehlers-Danlos
|
|
|
describe the heredity of the different types of ED
|
ED I, II and III - AD
ED IV - can be AD or AR |
|
|
which types of ED are known as classical ED and what exact defect is present?
|
types I and II
defect in type V collagen |
|
|
describe the clinical manifestations of ED I
|
- joint hypermobility
- skin hyperextensibility - increased skin fragility (cigarrete paper like scarring) - generalized tissue fragility, difficulty with wound healing |
|
|
describe ED II in relation to ED I
|
ED II is milder than ED I
- joint hypermobility is confined to hands and feet - minimal cutaneous involvement - tissue fragility rarely a problem |
|
|
What is the one clinical distinction seen in ED-III?
|
SEVERE hypermobility of all joints, usually without muscle or skeletal deformity
|
|
|
ED-IV is described as mainly what type of symptoms?
|
vascular
|
|
|
what are some clinical manifestations of ED-IV?
|
- spontaneous rupture of arteries
- spontanous perforation of bowel - prominent veins, easily bruise "gaunt, esthetic look" |
|
|
which type of ED is the most severe?
|
ED-IV
(Potentially lethal) |
|
|
which disease is often mistaken for child abuse by clinicians?
|
osteogenesis imperfecta I
|
|
|
what is synthesized less in OI type I?
|
less type I procollagen
(therefore less type I collagen) |
|
|
describe the collagen "population" in bone matrix in a patient with OI-I
|
- reduced type I collagen
- relatively increased type III collagen (therefore increased type III/type I collagen ratio) |
|
|
what are some clinical manifestations of OI-I?
|
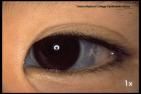
- multiple fractures before the age of 5 yrs. (decreased bone fragility after puberty)
- blue sclera - bowed extremities - osteopenia - otosclerosis/conductive hearing abnormalities |
|
|
what is the mutation type in OI-II and what gene is the mutation in?
|
point mutation in type I collagen gene (COL 1 A1 or COL 1 A2)
|
|
|
compare OI-II to OI-I
|
OI type II is more severe, can be lethal
OI type I individuals can live into adulthood |
|
|
what are some clinical manifestation of OI-II?
|
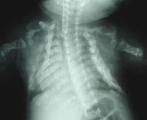
- multiple in-utero fractures
- crumpled humerus and femurs - limb avulsion during delivery - blue sclera - wormian bones |
|
|
what is the life expectancy of an individual with OI-II?
|
either:
stillborn or die as a young infant |
|
|
which disorder is often mistaken for osteogenesis imperfecta and has "telephone-receiver" bones?
|
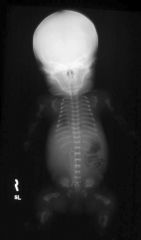
thanatophoric dysplasia I
|
|
|
Describe the X-ray differences between OI and thanatophoric dysplasia I
|
OI:
- accordian-type bones (rippled, wider) - multiple rib fractures thanatophoric dysplasia: - telephone receiver bones - narrow chest - no rib fractures |
|
|
what is the mutation in thantophoric dysplasia I?
what does this mutation result in? |
mutation in FGF 3 receptor.
this results in NEGATIVE bone growth activation |
|
|
describe the major abnormalities seen in an infant with thanatophoric dysplasia I
|

- impaired endochondral ossification, growth plate disruption
- macrocephaly - marked shortage of extremities - brain abnormalities |
|
|
what is the life expectancy of an infant with thanatophoric dysplasia I?
|
stillborn
OR death shortly after birth |
|
|
what is the hereditary pattern of most metabolic disorders?
|
AR
|
|
|
a patient with a deficiency of cystathione beta synthase has what metabolic disorder?
|
homocystinuria
|
|
|
what disorder is homocystinuria similar to and how can they be differentiated? (3 clinical symptoms)
|
similar to Marfan's syndrome
- homocystinuria has mental retardation, Marfan's does not - homocystinuria has downward lens dislocation, marfan's has upwards. - homocystinuria has thromboembolic events, marfan's does not |
|
|
what would the urinary and plasma tests show in an individual with homocystinuria?
|
urine - elevated homocystine
plasma - increased methionine, decreased cystine |
|
|
what eye manifestations would you see in homocystinuria?
|
downward lens dislocation
|
|
|
which metabolic disorder is characterized by a deficiency in glucocerebrosidase (beta-glucosidase)?
|
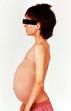
Gaucher disease
|
|
|
what is a Gaucher cell?
(where are they found, what is in them) |
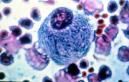
lipid-laden macrophages found in spleen, liver, bone marrow and lymph
- glucosylceramide accumulates in the lysosomes |
|
|
what are the three types of Gaucher disease and what is each type characterized by?
|
type 1. (chronic non neuropathic)
type 2. (acute neuropathic) type 3. (subacute neuropathic) types 2 and 3 are rare |
|
|
where is Gaucher disease type I most commonly found?
|
among the Ashkenazi Jews
|
|
|
clinical manifestations of Gaucher disease?
|
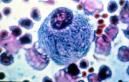
- hepatosplenomagaly
- anemia, bleeding tendancy, fatigue - thrombocytopenia - skeletal fractures (erlenmeyer flask fractures) |
|
|
which disorder is characterized by a deficiency of hypoxanthine-guanine-phosphoribosyl transferase (HGPRT)?
|
Lesch-Nyhan Syndrome
|
|
|
Lesch-Nyhan syndrome leads to an accumulation of what (2), which leads to an accumulation of what (1)?
|
an accumulation of hypoxanthine and guanine
leads to an accumulation of uric acid |
|
|
what are some clinical manifestations of Lesch-Nyhan syndrome?
|

- self mutilation (prefer to be in restraints)
- mental retardation - gouty arthritis - tophi: uric acid deposits (seen on pinna) - obstructive nephropathy |
|
|
which metabolic discorder is characterized by copper malabsorption?
|
Menke syndrome
|
|
|
what are some clinical manifestations of Menke syndrome?
|
- hair abnormalities
- cortical neuron loss - cerebellar dysfunction - "cherubic" face - seizures - tortuous cerebral vessels |
|
|
what is the prognosis for an individual with Menke syndrome?
|
progressively fatal, no cure known as of yet
|
|
|
in an individual with Menke syndrome, what would you expect the serum copper and ceruloplasmin levels to be?
what would you expect the erythrocyte copper levels to be? |
serum and ceruloplasmin levels will be low
erythrocyte levels will be normal |
|
|
what would calcium and phosphorous levels be in a patient with albright hereditary osteodystrophy?
|
calcium - low
phosphorous - high |
|
|
what leads to the abnormal Ca++ absorption seen in albright hereditary osteodystrophy?
|
a mutation in a G protein leads to an impairment of the cAMP response to parathyroid hormone. No response to PTH results in no Ca++ absorption
|
|
|
what other hormones are coupled to cAMP and are therefore impacted by albright hereditary osteodystrophy?
|
TSH
glucagon FSH LH |
|
|
which sex in albright hereditary osteodystrophy most often seen in?
which sex is it more severe in? |
most often seen and most severe in females (females affected 2-1 compared to males)
|
|
|
describe the IQ of an individual w/ albright hereditary osteodystrophy.
|
LOW (20-99 with a median of 60)
|
|
|
what are some other clinical manifestations of albright hereditary osteodystrophy?
|
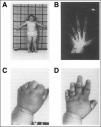
- obesity, rounded face
- delayed dentition - shortened metacarpals and metatarsals (4th and 5th) - diabetes mellitus and insipidus - short stature |
|
|
which disorder is a result of an 11p duplication and presents as hypoglycemia?
|

Beckwith-Wiedemann
|
|
|
what would an infant look like that was born with Beckwith-wiedemann?
|
Large (somatic gigantism)
- due to increased insulin, growth hormone and insulin like factor |
|
|
what are some clinical manifestations of beckwith-wiedemann?
|

- macroglossia
- omphalocele - ear lobe linear fissures |
|
|
if beckwith-wiedemann is not detected in the nursery what could occur?
|
nephroblastoma or adrenocortical carcinoma
(secondary to hemihypertrophy) |
|
|
which disorder is a result of a 5-alpha-reductase deficiency or a cytosol receptor defeiciency? (secondary to a genetic defect)
|
Testicular feminization (androgen insensitivity syndrome)
|
|
|
what is the inheritance of testicular feminization?
|
XLR
|
|
|
Which reproductive organs are present in testicular feminization?
|

testes present
uterus absent normal external female genitalia |
|
|
individuals with testicular feminization are phenotypically ___, gonadally ____, chromosomally ____ and overall are considered ____.
|
phenotypically female
gonadally male chromosomally male overall considered female |
|
|
a buccal smear taken of an individual with testicular feminization will come back as which sex?
|
male
|
|
|
what are some clinical manifestations of testicular feminization?
|

- infertility, amenorrhea
- sparse to absent pubic or axillary hair (due to no testosterone response) - breast development - shortened vagina |
|
|
A midline caudal defect that affects multiple systems, presents as potter facies, fused lower extremity and most often stillborn is?
|
sirenomelia sequence
"mermaidism" |
|
|
describe the clinical manifestations of the sirenomelia sequence
|
- fused lower extremity
- absent genitalia, bladder, anus - potter facies - lung hypoplasia - TE fistula |
|
|
what is "Potter syndrome," also known as the oligohydramnios tetrad
|

1. oligohydramnios
2. Potter facies (due to compression of face) 3. Pulmonary hypoplasia 4. Renal agenesis |
|
|
the following trait is often seen in the following disorders:
18p- trisomy 13 trisomy 18 cytomegalovirus |
cyclopia
|
|
|
describe the clinical manifestations and associations of cyclopia
|
- fusion of optic vesicles
- agenesis of corpus collosum - absence of olfactory tract - absent midline structures - cleft lip/palate |
|
|
What is the life expectancy of an individual with cyclopia?
|
most are stillborn
can survive up to 10 yrs |
|
|
what is the name of the disorder characterized by an aged "dwarflike" appearance and lax, wrinkled skin?
|
gerodermia osteodysplastica
|
|
|
what is the inheritance of gerodermia osteodysplastica?
|
AR
|
|
|
what are some other clinical manifestations of gerodermia osteodysplastica?
|
- lax, wrinkled skin
- aged appearance - osteoporosis - joint laxity - fractures - ptosis |
|
|
what is the life expectancy and intelligence of an individual with gerodermia osteodysplastica?
|
normal life expectancy, normal intelligence
|
|
|
what is the disorder characterized by excessive hair and abnormal teeth? (akin to "werewolf"...)
|
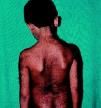
hypertrichosis lanuginosa universalis
|
|
|
what is the most common type of dwarfism?
|
achondroplasia
|
|
|
what are the two main defects in achondroplasia and what is the inheritance of achondroplasia?
|
1. mitochondrial oxidative system defect
2. FGF 3 receptor defect autosomal dominant with complete penetrance |
|
|
what are some complications seen in achondroplasia?
|
spinal cord compression
obstructive sleep apnia obesity |
|
|
what, skeletally, is wrong in achondroplasia
|
- broad and disorganized epiphyseal growth plate
- large cartilagenous epiphysis as compared to short diaphysis |
|
|
these three drugs, if taken by the mother, can result in?
Clomaphine Griseofulvin Valproic acid |
conjoined twins
|
|
|
what is the genetic problem in Angelmann syndrome?
|

15 (q11-13) deletion
genomic imprinting is seen here |
|
|
what are some clinical manifestations of Prader-Willi?
|
- hypotonia
- obesity (decreased perception of satiety) - hypothermia, hypogonadism - small hands and feet - delayed psychomotor development |
|
|
Which 2 disorders show a paternal age effect?
|
1. Marfan syndrome
2. Achondroplasia |
|
|
which disorder displays "Wormian" bones
|
osteogenesis imperfecta I and II
|
|
|
which disorder displays telephone reciever bones?
|
Thanatophoric dysplasia I
|
|
|
which disorder displays erlenmeyer flask fractures?
|
Gaucher disease
|
|
|
which disorder has a high risk of aortic aneurisms and therefore we prohibit acceleration/deceleration sports for these patients?
|
Marfan syndrome
|