Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
199 Cards in this Set
- Front
- Back
|
Insulin?
|
Insulin which treats diabetes is discovered in 1920s and shortly after ppl found it had an effect on diabetes but at that time there was a problem that you can only extract it from animals and if u used animal insulin to treat humans you got a side effect; second problem was the price because you got so little from each animal; in 1970s with development of moll technology ppl figured out how to use ecoli as host to develop human insulin so now it's affordable; used recombinant dna technique which is moll cloning.
|
|
|
If a protein wants activity, it needs a suitable
|
enviro and one critical condition is temp. For most human proteins we have suitable temp at 37 degree and if we get a fever, highest temp is 46 and if you get higher it's jeopardising life so we can use htis for PCR
|
|

|

|
|
|
original PCR
|
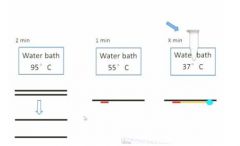
vvSo initial experiment: at the begininng you set up 3 water baths of the indicated temps. Then you take a tube and add the template dna and all the nucleotides as well as the primer into the tube and put it in the 95C bath for 2 min and then all your double stranded dna will be totally separated into single stranded dna.
Then you transfer the tube into the second water bath and cool it down to 55C. So here your primary structure is annealed to the single strand dna Then you take the tube out and put in the dna polymerase and put it in the 37C which is the best ttemp for the dna poliymerase to work so in here the dna polymerase will band to the primer site and start synthing the new strand of dna. The reason you added the polymerase at this step rather than the beginning is so that it isn't destroyed. They only get a small yield. Easiest way to get more is to repeat the process to double the yield. In each cycle you need to add dna polymerase in the third step and it's not a cheap compound. Each cycle is 5 minutes and if you do it 30 times that's 2.5 hrs so that's not a fun time so we use machines now. |
|
|
Scientists didn't think there was anything living in water nearly ______C so they just wanted to see if there was anything there and they found this bacteria so that means all its protein survives in hot water.
|

80 C
|
|
|
PCR components?
|
- template DNA
- dNTPs - Primers - Taq DNA polymerase |
|
|
brief PCR explanation?
|

|
|
|
After each mitosis cycle you're getting a
|
clone
|
|
|
YOu don't care if clones endup with exactly the same shape....
|
you care whether they have exactly the same genetic information
|
|
|
Which cycle doesn't produce clones?
|
meiosis
|
|
|
What's a common form of clones in plants?
|
grafting.
Most apples come from grafting Also using tissue culture techniques in a lab. |
|
|
omnipotent-ness?
|
All plant cells are omnipotent. In humans only the stem cells are omnipotent.
|
|
|
Farmers and gardeners have used vegetative cloning for_____________s of years`
|
1000s
|
|
|
dolly?
|

Take somatic cell from A and extract whole genome from that cell then you take egg cell from B and remove nucleus from there then you fuse the nucleus from A with egg cell from A which doesn't have the nucleus. After this, you culture it on a plate with medium which develops into an embryo which you transfer into a sheep which develops into a new lamb. So the new sheep has all the genetic info from A but it comes from the body of the sheep it was inserted into.
|
|
|
What kind of cloning will this class focus on?
|
cloning a moll or a piece of dna and is not cloning an org or a cell
|
|
|
plasmids
|
small circular dna structure naturally occuring in bacteria which can be replicated independently in the bacterial cell when it undergoes mitosis.
|
|
|
Bacteria always have plasmid because
|
it needs it because it has antibiotic resistent gene that makes ecoli resistent to certain antibiotic
|
|
|
The first RE found in Ecoli was
|

EcoR1
So for this enzyme, it scans along the dna double helix and when it finds a specific sequence as shown in the blue here, it stops and cuts the double strand and makes two smaller molls |
|
|
What is a palindromic sequence?
|
well when you read it in the opposite direction, you read the same sequence
|
|
|
Each restriction enzyme can only work in one
|

in one way
|
|
|
with plasmids the function of ligase
|
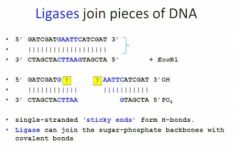
is the same as in dna replication which is to draw the dna fragments together as one dna fragment.
At the beginning here vv we have a double strand dna and we use EcoR1 to cut this double strand dna because it has a restriction enzyme for EcoR1 and then you have two small moll that look like that. So we can see here for the sticky end of this it's totally complementary. When they're complementary, they're compatible sticky ends, so they'll tend to create hydrogen bonds but without ligase they'll break again later. Ligase provides an opportunity for them to be very close. |
|
|
Only when the sticky ends are compatible
|

can they bond
|
|
|
ampicillin resistant ecoli experiment?
|
In one tube we have a plasmid and we add EcoR1 as the RE. Because this plasmid has been engineered to have only one RE for EcoR1 so we only cut this plasmid at one position and make the circular dna structure into a linear dna structure.
Then in another tube, we have our human dna there and also added EcoR1 in that part. The human genome is very big and probably has many EcoR1 sites so in the end what we have is there are many dna fragments cut by the EcoR1 and on the end of each fragment there's a sticky end for Ecor1 because they are all cut by EcoR1. Then we purify the linear plasmid from Tube A and then we purify a piee of dna fragment from tube B and then we assume that in the tube B the fragment that we have we extract from there is having the insulin gene for humans so we assume this is the blue part here and you can in fact purify any part from solution. After we purify these two part together and put them together in one tube it will ook like this, you have linear dna structure from tube a and you have small piece of dna from tube b. So right now if we add the ligase in this tube, the ligase will join the sticky end together and make these two fragment binding together and forming a bigger circular dna structure like that and in this case we successfully insert a piece of human genome into a plasmid and this what we call a recombinant plasmid vector. So next, you just need to transfer this new plasmid to Ecoli. So you transfer this thing to Ecoli on a plate that's containing ampicillin so only the ones which contain the plasmid can grow. |
|
|
Electrophoresis separates molls by
|
charge and size
|
|
|
DNA and RNA have uniform __________________ charge
|
-ve
|
|
|
DNA ladders are
|
mixtures of fragments with known size
|
|
|
using diff primers
|
gives you diff sizes.
diff primers gives you diff sequences |
|
|
difference between sequencing and PCR?
|
- Need taq polymerase, DNA template, primers (only one primer unlike PCR), dNTP, ddNTP (unlike PCR). There's no 3'oh group on ddNTP and we have known from dna replication that 3'oh group is very important for elongation... without it you can't have the next nucleotide binding to elongate the strand... so in that case whenever a ddNTP is integrated into a growing band, the band will stop there and not be elongated any more because there's no 3'oh group. The next nucleotide will band to the 3'oh group... if there's no 3'oh group that reaction can't happen so it stops there. ddNTP is a very similar structure to dNTP.
|
|
|
Sanger DNA sequencing: ?
|
- Put that stuff in then what happens in the machine? Similar to regular PCR process. Heat up tubes to 95C and then double strand is separated into single strand, then you cool to 55C so the primer is annealed to the single strand> After that you start with the elongation step which is 72C and in this step the taq polymerase will work from your primers and to elongate the primer structure according to the template.
|
|
|
Sequencing industry is developing very fast....?
|
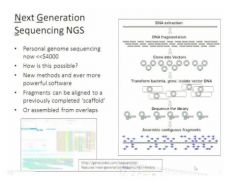
This is another sequencing technique right now. This technique started in 2008 and it became available in 2010. This technique can produce a huge amount of data shortly and cheaply. Human genome project was happening from 1990s to 2000s which cost about 3 billion $ to finish. Here by having this technique we can do this in about 2 weeks. Some company is announcing that it only costs several thousand dollars to sequence the human genome. So this provides the possibility to sequence every person's genome and also provides the possibility to sequence every organism, but right now it's so easy and so fast. This technique does have problems though that it can't target any gene, it has to be the whole genome (unlike Sanger). Also this method relies on the computer to analyze because you can't analyze it yourself because of the huge amount of data.
|
|
|
central dogma of molecular biology means
|

that the genetic info that is gong to encode proteins and other regulatory sequences that permit the formatino and functioning of a cell is contained in the dna that`s in the nucleus or in the nucleoid region in a euk cell or a prok celk. This is the library copy, it doesn`t actually have a direct functioning application to the cell because it has to be transcribed into an rna moll so rna and dna are comparable in nucleotide sequence so they contain the same kind of info but they`re differently regulated in terms of activity and stability so it`s going to be copied into an rna moll and this is going to be encoded into a polypeptide, the protein which has the structural or functional role in an enzyme.
|
|
|
where does 3`oh and 5`phosphate come from?
|
Sanger's sequencing is a fundamental method for determining the nucleotide sequenc ein a region of dna and it now has applications for sequencing rna as well. Here we have a standard nucleotide. In a nucleotide you have a sugar and this sugar is ribose. A dna base in this case it's cytosene which is a pyrimidine and it has a phosphate so when yo unumber the attachments to a sugar they're numbered from the carbon. So the 3' Oh comes from this and the 5' phosphate. With RNA you have a slightly different organization. For rna, you have a slightly diff organization but the important thing is that although in rna this is an h instead of an oh, both of them have a free 3'oh which is critical for dna polymerase and for rna polymerase to continue to add basesvvv
|
|
|
When can you polymerize and when can you not?
|
So here we have a cartoon of a double stranded segment of dna and so you can see that one end has the free 5' phosphate and the other end has the free 3' oh and if you have that you can polymerize and if you do not, you can notvv
|
|
|
because hydrogen bonds are relatively weak and numerous they can be separated by
|
heating the dna
|
|
|
ddNTPs?
|
How does this get used for sequencing? No wwhat we're going to do is to modify, use a mixture of nucleotides so that the vast majority can continue the dna polymerase reaction until the end of the extension cycle, but about 1 in 1000 are going to be special and what these are going to be is di deoxy nucleotides so ther is no free 3'oh this has been taken out so that as randomly some individual nucleotidse are incorporated polymerizatino is terminated at that nucleotide base and these are called terminators and so what's going to happen here is for eg a labled atp so it's got no possibility of continuing the polymerization reaction but it will have a tag that says here I am, so the poollymerization continues and then at some point at a random T because it's an AT base pairing you will have that particular strand is no longer going to be able to extend so if you have then a small number of these di deoxy atps, polymerization will start and it occurs at about a rate of 1000 base pairs per minute and it will continue continue continue and it will hit one of these di deoxy atps that has been tagged in a way that we'll be able to see it and that polymerization will cease but others going on at the same time in the same reaction from the same time are going to be able to continuevvv
|
|
|
Agarose is
|
a simple carb with 5 carbons water solutble at high temps and then it forms a gel sort of like gelatin except it's a carb gel instead of a protein gel and then it cools and the conc of agarose in the gel is going to determine the pore size (relative resistance to flow)
|
|
|
What makes dna and rna negatively charged?
|
the phosphate attached to it that has extra oxygens
|
|
|
Electro phoresis for dna sequencing
|
because we are trying to differentiate fragments that differ by a single base pair not by a thousand base pairs, you have to use a slightly diff gel that has a much finer pore size
|
|
|
in electrophoresis, DNA is visualized by
|
specific dyes
|
|
|
how do ddNTPs work in sanger sequencing?
|
there's going to be a tagged terminator that is going to interrupt dna synthesis at the complementary base pair for the nucleotide in question. So here if you're adding ddNTP you are terminating the sequence at the corresponding thymine (residue).
|
|
|
electrophoresis procedure?
|
you 've tagged your di deoxy atp, you run it all on a gel, at this point it's invisible, but you have separated the fragments one base pair after another by single base pair amounts and then you take the gel and overlay it with xray film. As the P32 undergoes a fission reaction it lets off a high energy gamma ray which exposes the gel.
|
|
|
You get a lot of sequence info in electrophoresis
|
for the first few hundred base pairs close to the dna primer and then relatively you see fewer and fewer and in order to continue to query that sequence, you generate another sequencing primer here and you do it again. This is called primer walking.
|
|
|
in the early days you could read up to
|
300 bp if you were lucky
|
|
|
later on the sequences were tageed with
|
a fluorescent primer instead of the 3'OH and what this allowed was to run all of the reactions together so you could do all of the sequencing in one tube instead of four tubes and you could have that combined product and run it thru a gel that at the bottom had a fluorescence reader so that it could tell you which colour of fluorescent tag was being processed (coming off the bottom) at any one time) This allowed for long reads.
This allowed for long reads. Longer reads are important because longer erads means fewer overlaps, fewer overall sequencing reactions and more fidelity. Increases in the affinity of some of the tags and the affinities and efficiencies of the thermostable dna polymerase now allows us to read up to a thousand base pairs for a single sequencing reaction. |
|
|
Sanger's seq is important...
|
even today even though we now have many other techniques for reading dna sequencing because it reads the template sequence directly and allows for high fidelity and for the ability to query a particular dna seq.
|
|
|
Other kinds of sequencing\??
|
Some of you may have heard of next generation sequencing which is often an entire genome which is fragmented into relatively short pieces and multiple overlapping fragments are sequenced often a hundred million fragments which are overlapped overlapped overlapped to permit figuring out what the consensus sequence is. Sanger's is an important first step because it provides an unambiguous read of the backbone. So if you are doing multiple sequencing then you will have multiple overlapping fragments and it is the consensus that it gives you the seq, but because there are so many copies of everything it is much more complicated, there is a lot more bio tech processing that has to be done to make sure that you are getting an unambiguous result.
|
|
|
dna sequencing detection depends on the
|
labeling method
|
|
|
in the 1980s you could read?
|
300 bp readable (32p)
|
|
|
1990s you could read
|
500 bp readable (fluorescent tag)
|
|
|
Nowadays you have sequencing software for
|
alignment and annotation
|
|
|
Before info in dna can be used it needs to be
|
transcribed in a process that is analogous but not entirely similar to replication
|
|
|
what is a gene?
|
a heritable unit of information
|
|
|
The part of a gene that mendel was studying is
|
the transcribed region which is the actual code for the gene product and it is the part which is eventually translated
|
|
|
upstream of the transcribed region towards the 5'end is
|
some important nucleotide seqs. The most upstream one is called the promoter and then there is a series of regulatory seqs which we now know are extremely important for determining how often a gene is transcribed.
|
|
|
If it can't be transcribed
|
then it can't be translated.
|
|
|
And at the 3' end there
|
there's a terminator
|
|
|
the promoter
|
allows the assembly of the rna polymerase that's going to copy the dna into rna
|
|
|
ther termintator
|
causes diassembly of the machinery so after the rna polymerase binds to the promoter and this is its ability to bind (controlled by the regulator) it transcribes and then falls off
|
|
|
and the primary transcript here is the rna and the transcribed region is the part that encodes the info for the translation product but this is going to vary dependin on what kind of cell we're talking about
|
and the primary transcript here is the rna and the transcribed region is the part that encodes the info for the translation product but this is going to vary dependin on what kind of cell we're talking about
|
|
|
gene expression requires
|
transcription and translation
|
|
|
promoter and regulatory sequence
|
binding sites for rna polymerase and accessory proteins
|
|
|
transcribed region
|
coding region
|
|
|
terminator
|
releases RNA polymerase
|
|
|
Difference between prok and euk transcription and translation?
|
So in a prok cell, you will recall that the nucleoid region in that cell is within the cytoplasm of the cell so that there`s no barrier between the dna and the translation machinery (the ribosomes). Much more complicated in euk cells because the transcription is occuring within the nucleus but the translation is occuring in the cytoplasm.
|
|
|
Why the difference between transcription and translation in prok and euk cells?
|
Why would euks do this? Because it provides an dextra level of control and organization so we can get more subtleties in the translation products for a euk sequence compared to a prok sequence. As soon as a prok gene is transcribed it si already available for translation, and in contrast in a euk cell there is a spatial delay and a temporal delay between transcription that allows for some processing in the nucleus so there can be differential processing of certain kinds of transcription products so you can get more than one kind of info out of a particular gene sequence and then after modification the mature messenger rna is going to be carried out into the cytoplasm and that's where it's going to be translated. So in euk cells transcription occurs, in the nucleus translation occurs, and in the cytoplasm and in contrast in proks all of this happens in the same place and essentially at the same time.
|
|
|
messenger RNA is abbreviated
|
mRNA
|
|
|
All cells: genes are transcribed into _______ __________ before translation into proteins by the ribosomes
|
messenger rna
|
|
|
the promoter region is important for
|
binding the rna polymerase to the double stranded dna and prepare it for unwinding using factors including the sigma factor because just like dna replication rna transcription requires that the double stranded dna be separated so that the nucleotide bases be exposed.
|
|
|
In order to not lose info , the transcription site is going
|
is going to upstream 5' of the translation start but then after it carries thru the dna it will end at the terminator
|
|
|
promoter region is
|
recognition site for sigma factor and RNA polymerase
|
|
|
DNA is unwound to create
|
the open complex
|
|
|
Trnascription start site is slightly upstream of the
|
translation start
|
|
|
transcription ends at
|
the terminator site
|
|
|
rna polymerase binding to the promoter site with accessory proteins including the sigma factor which are going to open up the dna double helix to allow for enzyme activity so it requires a polymerase as with dna replication and in this case it's rna polymerase because rna is going to be transcribed from the original dna seq
|
|
|
|
diff between dna and rna
and stability of rna? |
because for dna you have four bases and for rna although you also have four bases, instead of thymine you have uracil, uracil is also a pyrimidine and it base pairs with adenine, it forms the same 3 hydrogen bond coordinate bonding but it is an older kind of nucleotide and it is somewhat less stable. The stability of the rna product then is somewhat lower than the dna product and this is important because if it were not that rna were somewhat more ephemeral when you transcribed dna into rna then that gene transcription product would be maintained in the cell for a long time so it's important for messenger rna not be stable, it has to be able to be turned over so the cell can not only turn on gene function but also turn it off again.
|
|
|
unlike dna replication, we are only going to be transcribing
|
one strand. Only in the 5' to 3' direction, so there's no okazaki fragments in the transcription process.
|
|
|
the open complex allows
|
the access of rna nucleotides into the transcription site
|
|
|
RNA polymerase dissociates at the terminator, and is available to
|
transcribe a new gene (or the sme one again)
|
|
|
In prokaryotes, as soon as the mrna as been transcribed from the dna,
|
the ribosomes can land on that primary transcript even before it is complete and they can already be translating protein so it's a very fast and efficient process and this is part of the reason that prok life cycles can be very fast, because there's no waiting here.
|
|
|
eg of quick rna synth in proks?
|

immediate landing of the ribosomes onto the transcript and they can begin peptide synth even before the message is complete so you can see here we have the earliest transcribed mrna and ribosomes that are working along that mrna and are translating it immediately as it is transcribed
|
|
|
In euks there are exons which are
|
the part that';ll be translated
|
|
|
introns have to be
|
spliced out by a euk specific regulatory mechanism
|
|
|
There are recognition sequences that these protein machines can land on so that
|
these parts are precisely spliced out and then the mature messenger rna is transcribed
|
|
|
So why would you do this intron exon thing?
|

It seems like an awful lot of work and somewhat risky but what's happening here is that if you have a single primary transcript in some cases you may choose to remove all the introns, if you're a cell. In some cases all of the introns will be removed and you'll have a full length transcript, but in other cases you have differential processing at this stage that allows the cell to encode more that one kind of translation product for a given transcript so this allow the euk genome to have tremendous amount more flexibility.
|
|
|
poly a tails?
|
other thing happening here is that the 3' end of a euk transcript is modified at this stage by adding what's called a poly a tail so this is a polyadenilation chain which is added here and the poly a chain has some very important features, it is added to a recognition site here and it is in fact the gate keeper which allows the mature message to leave from the nucleus out into the cytoplasm so the poly a tail then is the cellular message that allows the mature message to leave from the nucleus and get out into the cytoplasm
|
|
|
The poly a tail is then the
|
cellular message that allows the mature message to leave from the nucleus and get out into the cytoplasm so there's a signal site and this signal site as you can tell is in rna cause there is uracils rather than thymidines and so what's going to happen here is that after the message has exited into the cytoplasm then translation can take place so you have the message with a poly a tail, this can be translated translated translated, however at the end of the translation activity cycle, the poly a tail is removed and the poly a with the removal of that poly a tail then the message is targeted for depolymerization and for recycling.
|
|
|
eg of regulation
|
there are regulatory seqs that are important for determining the relative activity of binding of the rna polymerase complex.
There are regulations in the accessibiliyt to splicing and there are alternate splice sites so you can get more than one kind of protein product from a message. Then there's regulation as the mrna is taken out into the cytoplasm because it has to interact with a protein complex to get there and then finally there's regulation by the timing and relative activity of poly a degradation that is going to turn off translation |
|
|
There are no introns in
|
proks
|
|
|
In proks the primary transcript is translated directly
|
into protein
|
|
|
In euks it's more complicated than in proks (rna synth) because
|
IN euks it's more complicated cause the primary transcript is subject to rna splicing and processing which allows for differential regulation of the message so tha tmultiple kinds of transcripts can be translated from a single message and in particular the introns of some important internal regulatory sequences. There's also the addition of a poly a tail which is important for getting the transcript out of the nucleus where it si formed but cannot be translated into the cytoplasm where it can be translated and regulated.
|
|
|
Summary of dna transcription:
|
This is the summary for dna transcription, you have the double stranded dna, you have the landing of the rna polymerase on the promoter region , you have the opening of the double stranded dna helix, you have the copying of one strand and one strand only into rna dn then in a a prok you can have immediately translated product and in a a euk you have to have additional processing and moving of that primary rna out into the cytoplasm.
|
|
|
ribosomal machine?
|
It's a two part machine made largely of ribosomal rna subunits and these are going to be decorated with proteins that provide functional information.
|
|
|
RNA polymerase dissociates at the terminator and...
|
is available to transcribe a new gene (or the same one again)
|
|
|
Ribosomes are rna...
|
rna protein machines
|
|
|
ribosomes come in two...
|
two subunits, the large subunit and the small subunit and these are very diff functions
|
|
|
(general overview of ribosome function cycle starting at end of translation) When translation is over...
|
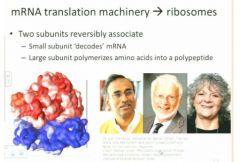
ribosomes dissociate so that they get reused one association for each translation event so what's going to happen here is that the small subunit shown here in blue is going to be where the message is bound reversibly and this is going to be where the decoding is for the information that's in that message and is the large subunit that's going to be involved in polymerization of the protein product.
|
|
|
in depth understanding of ribosomes?
|
all this now is understood extremely well with high structural detail. About four years ago the final crystal structure was solved in high resolution so now we have very precise information about how this is done. And as many things go in science, this was not an individual effort but was a collaboration and it was an international collaboration.vvv
|
|
|
The different ribosome subunits are called
|
ribosomal cores
|
|
|
there are many _________ associated with each ribosomal core
|
proteins
and each one has functionality |
|
|
Back when ribosomes were first evolvinb
|
there would not have been proteins, this entire catalytic cycle would have just been the ribosomal rnas.
|
|
|
there are parts to these ribosome subunits and these are homologous but not absolutely identical in bacteria and in euks
|
the bacteria ribs are somewhat smaller than the euk ribs
|
|
|
Svedverg?
|
S in this case stands for Svedverg. Back when cell biologists and physiologists were trying to understand how the relative size of diff machines, they had to develop a methodology for doing this and Sved came up with the idea that if you centrifuged a suspension fo small aprticles, the rate at which they moved thru the suspension when accellerated by a million gravities which we would generate would be consistent measure of sedimentation. So S is also for sedimentation and it is not an absolute 1 to 1 correlation with molecular weight of the subunits.
|
|
|
size of proks ribs?
|
So for the proks, the overal size of the prok rib is 70 s but it is divided into the 50s large and 30s small so you can't just add up the small pieces to get the big pieces.
|
|
|
size of euk rib?
|
The euk rib is somewhat larger both in the size of the rib rna and also in the number of proteins involved
|
|
|
many of these ribosome proteins are involved in
|
are involved in how the mrna docks and interacts , how the protein product is moved thru the machinery so S is for svedberg and this is hwo many of the small components in cells are defined for sizevvv
|
|
|
S is for
|
Svedberg, which describes sedimentation rate
A substance with a sedimentation coefficient of 26 S (26x10^13s) will travel at 26 microns per second (26x10^-6 m/s) under the influence of an acceleration of a million gravities (10^7m/s^2) |
|
|
TRNA etc?
|
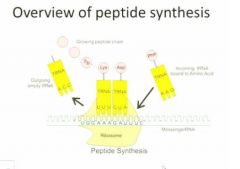
An overview of what we're going to be talking about here. We have the large subunit on top and the small subunit on the bottom and here they're shown as a single piece. You see that the mRNA is docking on the small subunit of the ribosome and hwat'll happen is that transfer rna (special kind we haven't talked about yet) will act as a decoding machinery that can recognize the mrna thru base pairing at one end and they have a covalent bond to a particular amino acid at the other end so for each codon you'll have a separate transfer rna andso you have a particular transfer rna for each type of codon and these are covalently linked then to a particular amino acid. These are highly conserved
|
|
|
cartoon of a more realistic veiew of a transfer rna?
|
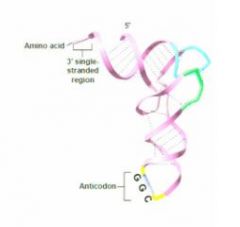
|
|
|
inter strand base pairing of tRNA?
|
They have homologous sequences that allow for internal inter strand base pairing that allows them to fold up into this particular shape
|
|
|
Each a mino acid has it's own
|
transfer rna
|
|
|
Why is the structure of the rnas highly conserved?
|
because they have to fit properly into the large subunit ribosomal binding sites
|
|
|
Messenger rna binds to
|
the small subunit before the large subunit docks
|
|
|
start codon?
|
is always the same for every protein product and it's always going to encode thiamine.
|
|
|
upstream region of rna
|
important for stability of this docking
|
|
|
P site
|
site where the peptide bond will be formed
|
|
|
a site
|
where the next amino acid bound to its transfer rna is going to dock to the next codon on the mRNA.
|
|
|
e site
|
the exit site where the empty transfer rna that's given up its amino acid to the peptide chain is going to leave the large subunit.
|
|
|
moving from one amino acid to the next?
|
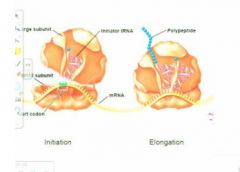
What we're going to have then here is what we saw before so you've got the initator transfer rna. Now we have the addition , the entry o ft the second trasnfer rna carrying the next amino acid residue for the polypeptide and what' shappening heree is that there's catalysis that is going to form the peptide bond between the existing, or in this case the first amino acid with the next, subsequent amino acid, so having had initiation then we have a series of stepwise additions of single amino acid residues, one by one for each anticodon along the messagevvv
|
|
|
moving up to termination of rna?
|
So we've had initation, we now have elongation and proteins can be from short ones a couple hundred amino acids, up to thousands of amino acids, so the elongation stage can be extremely extended and then eventually, the message will hit the stop codon (will talk about in a bit) and at this point I t will trigger dissociation, release of the polypeptide, release of the message, and this is the process of terminationvv
|
|
|
n is the
|
amino terminal
|
|
|
c is the
|
carboxy terminal
|
|
|
there are ____________ amino acids
|
20
|
|
|
Who figured out that a codon is 3 base pairs?
|
Nirenberg
|
|
|
many amino acids are encoded by more
|
than one triplet
|
|
|
all protein products when they are initially synthesized begin with
|
thianine
|
|
|
What happens at the first base pair in the triplet
|
is going to be the most important part of the decoding
|
|
|
for most amino acids, the first two places in the codon
|
are the most important for determining the aino acid that's going to be encoded
|
|
|
the third amino acid
|
is often not so important and what this is called is the wobble hypothesis
|
|
|
thianine is also called
|
AUG
|
|
|
what are the 3 stop codons?
|
uag, uga, and uaa
|
|
|
upstream includes the
|
the promoters and regulatory regions
|
|
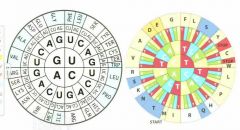
similar colours have
|
similar chemistry
this wheel is called sunflower versions |
|
|
if you have second base pair mutations
|
you may have a change in the actual amino acid but not in the chemical specificity
|
|
|
even if you have mutations in the message
|
you can still encode a potentially useful protein product
|
|
|
The polarity of the message 5' to 3' is directly
|
mirrored in the polarity of the amino acid chain that it encodes.
|
|
|
what determines the structure and folding of the proteins?
|
the side chains
|
|
|
polarity of side chains?
|
some are polar, some aren't. some are charged both positive and negative
|
|
|
cysteine?
|
there is the amino acid cystein which has the capacity to form covalent bond between cysteine which can provide a region of high stability and cross linking. (polypeptides)
|
|
|
two main important side chain interactions of polypeptides?
|
alpha helices which are involved in certain kinds of functions. These are going to be important particularly for membrane spanning and transmembrane proteins.
|
|
|
polypeptide hydrophobic interactions
|
There are also a large number of hydrophobic interactions. These hydrophobic interactions are based on Vander Valls forces so these are associated with regions of primary protein structure wher all or most of the side chains are hydrophobic so these tend to repel water and they tend to be in the middle of the finely folded protein.
|
|
|
disulphide bridges and ionic bonds in polypeptides
|
There are disulphide bridges which are important for stability of certain cross links and occasionally there are ionic bonds between positively and negatively charged side chains which are important for other kinds of folding.
|
|
|
tend to find in polypeptides?
|
In a fully folded protein then what we tend to find is hydrophobic intereactions on the inside of the protein that exclude water and allow for stability and on the surface we tend ot find charged regions that are important for protein ineteractions between diff members of a complex.
|
|
|
Proper protein folding is essential for
|
proper protein function
|
|
|
Evidence of protein folding stuff?
|
We have two kinds of evidence for this: we have misfolded proteins due to mutations and we also have misfolded proteins due to prions diseases. Prions are normal proteins but they have for various reasons misfolded into a stable conformation that is nonfunctional; they have the capacity when they associate with a ordinary protein of the same type to cause the misfolding of that protein so it is a protein folding problem that leads to product wasting disease, tha tleads to alzheimers disease, that leads to a whole series of very difficult to treat problems and here it si not the primary amino acid chain it is not a defect in folding but in the fact that a misfolded protein can change the tertiary structure
|
|
|
Polypeptide chains fold based on the properties of the
|
amino acid R-groups
|
|
|
each amino acid is linked by
|
a peptide bond that was formed in the large subunit of the ribosome
|
|
|
possible secondary structures?
|
secondary structure which is a helix or a secondary structure which is called a beta pleated sheet and these tend to go back and forth, they have a different kind of stability.
|
|
|
protein pla ces where there is no defined secondary structure?
|
regions which are called coils
|
|
|
alpha helices and the beta sheets are typically involved in
|
stabilizing the structure of the protein whereas the loops typically are more involved in dynamic interactions with other proteins
|
|
|
Not all proteins are individual for their function and
|
sometimes you have multiple peptides working together to produce a functional subunit
|
|
|
different protein structures?
|
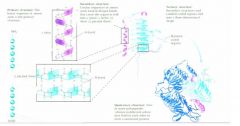
|
|
|
So the quaternary protein structures are
|
the multi moll machiens which are required for higher order function.
|
|
|
Actin is the most
|
abundant protein in our cells.
|
|
|
Actin is involved in
|
the contraction of muscles. It's involved in many features of motility and euk cells and related proteins in prok cells.
|
|
|
What are the two ends of actin?
|
The pointed end and the barbed end.
|
|
|
the pointed end of actin has
|
the binding site for functional molls which include atp and calcium and large numbers of these subunits and large numbers are going to be able to asociate with each other in order to form the quaternary structure filaments
|
|
|
What areas can you see in this diagram of actin
|
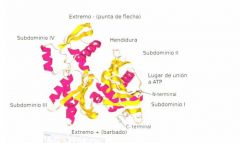
regions of secondary alpha helices, you have regions of secondary beta sheets. you've got the loops which will be important for interactions between the subunits, you've got binding pockets for co-factors and all of these together are going to form a structural moll
|
|
|
What are the two important kinds of quaternery structure?
|
There are structures like this for actin and microtubules, where you have a repeating polymer of the same kind of subunit over and over and then proteins which interact with these sides of these filaments provide extra functionality and extra specificity for targeting these proteins.
We also have enzymatic quaternary structure, and an example is hemoglobin which has four subunits, in this case they have to work together cooperatively to find oxygen and to release it as necessary. |
|
|
If you have a change in the third base pair, it's called a
|
silent mutation
some large proportion of random mutation will have no effect at all |
|
|
semi conservative protein mutation?
|
when you go back to the wheel and look at the semi redundancy of the code you can see that it's possible to have changes from one amino acid to another amino acid which are semi conservative so instead of having in this sense a mutation that changes from one chemical amino acid to another , you can have relatively small change so that you have some conservation of function even though you've had a change in the conservation of structure
|
|
|
kinds of messense mutations
|
premature terminations leading to truncated proteins
frame shift mutations: insertions or deletions into the message |
|
|
When we study the underlying features in physiology, in agriculture, in engineering, it almost always comes down to
|
identifying and manipulating genetic seqs
this can span everything from chem engineering to health sciences |
|
|
Potential problems with GMO are related to
|
downstream or peripheral issues with selective pressure to maintain the preponderance of the gmo and the other problem is in outcrossing between a gmo food crop like canola and wild meaty relatives that can potentially then acquire the same kinds of resistance.
|
|
|
applications for molecular biology
basic science? |
- genetics, biochem, taxonomy, medicine agriculture
|
|
|
applications for molecular biology
biotechnology? |
- cloning, GMOs, artificial protein production
- xenotransplantation, crop improvement |
|
|
what can cause gene mutations?
|
natural things like UV light, certain chemicals that are dna damaging and it can also be caused by certain viruses
|
|
|
dna mutations causing damage?
|
random mistakes in dna replication typically cause damage to the org in experimental situations we can use that kind of damage to an indiv gene to understand the function of that gene. So it's like taking indiv pieces of a car out to see what each piece does. So random mistakes in dna replication then are a powerful experimental tool that has allowed investigators to study aspects of gene function.
|
|
|
Beadle and Tatum?
|
Beadle and Tatum in 1950s used this kind of information to dissect the argenine biosynthesis pathway of ?a tractable model system ???To understand that there are multiple steps in argenine biosynthesis . So they created a suite of mutants that were unable to grow without the addition of argenine, hypothesized that these mutations were involved in the pathway, discovered that there were various kinds of the mutations and formulated in the end the hypothesis that said that a gene seq was responsible for encodinga polypeptide.
|
|
|
problems as a result of biochemical knowledge?
|
One of the downstream problems is because we've developed these techniquesk, we've eneded up in the unfortunate situation where we're using chems for our daily lives like for the production of food and animals we use an awful lot of antibiotics and have led to problems such as the development of microorganisms that are resistant to almost every kind of antibiotic we can give them...
A particular problem right now in hospitals is what we call m????? And so here we have an org that is resistant to a drug that used to be a powerful drug and it's no longer therapeutically useful, so the questin o is how likely is this to have happened. penicillin story |
|
|
penicillin story?
|
A particular problem right now in hospitals is what we call m????? And so here we have an org that is resistant to a drug that used to be a powerful drug and it's no longer therapeutically useful, so the questin o is how likely is this to have happened. Back in the 1940s when pennicilin which is chemically related to methicillin , there was a great biosynthetic effort to create peniccilinn which had been understood for decades to be an important anti-bacterial drug, but they didn't have very much. In fact all the peniccillin on the planet wasa a single gram. They had a human patient who had scratched his hand and had developed a bacterial infection in his blood called septosemia and at the point where the first peniccillin therapy was attempted, this patient was comatose near death, they gave him a dose of penicillin and over night he recoverred, came out of coma and was hungry. But they didn't have very much, only used a portion, they used the remainder of it over 3 days and in the very short time for a naïve bacteria, the bacteria developed a resistance and he died in a week and a half. So one of the important things to think about is that if you use a selective agent like a drug, if you use a therapeutic agent, you inevitably develop a resistance to that agent because bacteria and microorganisms grow very quickly and the faster they grow the more chances they have to have mutations during dna replication and the more chances they have for selection of resistant mutants.
We've used compounds like penicillin, methiciling, ampicillin, all the time now for lots of reasons, many of our pathogens have developed a resistance against them. |
|
|
in ecoli you get about _____ generations in a day
|
72 generations in a day
This is about as many generations as humans have had for a couple thousands of years |
|
|
Granny smith apples?
|
Granny smith had an apple farm in South Africa and one branch had unusual apples and all the granny smith apples we have today are direct descendents of a single branch of a single tree. The point here is that cloning per say may or may not involve genetic methods and it may or may not be GMO
|
|
|
genetic cloning uses molecular
|
techniques and tools
|
|
|
Enzymes manipulate dna
|
directly
|
|
|
most important enzymes
|
are restriction endonucleases which recognize the specific base pair seq in the dna and cleave it in a site specific manner and these have allowed researchers to specifically target and cut dna at defined place so it's possible to separate and re ligate (combine) it in defined ways So the RE cut in specific places. Ligases join seqs back together and polymerases are important for replicating dna and are also important for sequencing.
|
|
|
Gel electrophoresis separates
|
dna or rna or proteins depending on the kind of gels and the kinds of methodologies. It separates them by size based on a common charged and the fact that these are being forced electrically to migrate.
|
|
|
Cloning vectors?
|
Plasmids, extra ???nucleosome?? Dna which can be used as artificial chromosomes to host a seq of interest for replication
Dna seq is used to confirm that the manipulation of choice have occurred as expectedvvv |
|
|
enzymes
|
polymerases, ligases, restriction endonucleases
|
|
|
cloning vectors
|
plasmids to artificial chromosomes
|
|
|
enzymes like REs naturally
|
occur in bacteria and appear to have evolved as a protection method against viral dna
|
|
|
So like everything bacteria have
|
viruses. The viruses can attack the bacterium and if the bacterium can produce an enzyme that cuts the viral dna then the dna can be disabled.
|
|
|
The first RE to be discovered was called
|
Ecor1 and it recognized gaattc, we always talk about this in the 3' to 5' direction and one of the interesting things about REs is that they are palindromic and what that means is that you can read them in the forward direction or in the complementary strand they are identical forward and reverse.
|
|
|
All of the RE names relate to
|
the orgs from which they were originally isolated. So ecor1 comes from ?????? Ecoli which is a common gut bacterium.
|
|
|
Diff Res and there are hundreds now all recognize palindromic seqs but they recognize
|
specific seqs
|
|
|
Hundreds of different REs have been isolated from
|
various bacteria, recognizing specific base sequences
|
|
|
how do REs bind to the DNA?
|

The way they do this is that they're dimers, they bind around the dna so here we are looking down the dna double helix. So you hve two enzymes of two monomers of hidiIII which are forming a dimer so here we have a case of quaternary association used for function. They are binding to andable to recognize the internal seq of the base pairs and cut in a precise wayvvv
|
|
|
If you take sticky ends, mix them together and control the temp properly, you can allow
|
reannealing
This is not going to be very stable because the number of hydrogen bonds here is very low but if you can manage to do that then you can add a dna ligase to reform that phosphodiester bond and regenerate a double stranded dna so what's happneing here is you must have a 3' oh at the end of the guanine and a 5' phsophate at the adenine and what's going to happen then is with the ligase with energy from ATP si your'e going to be able to reform that phosphdiester back bone. |
|
|
can you join hindIII sticky ends with ecor1 sticky ends?
|
no
|
|
|
lack z experiment?
|
So sofar this seems to be okay except that now we've got diff sites and how are we going to put things together that have unlike sites? At the end of the early 1990s, researchers at the U of California took a plasmid and engineered it very carefully so that the plasmid which is shown here as the circle had a resistant marker for ampicillin so a bacterium tha thad that plasmid would be able to grow on ampicillin. It had an orgiin of replication so that it could replicate itself. |But most importantly it had two features here. One was called the lack promoter and the lack z enzyme that would allow it to form a blue compound when it was grown on a particular substrate but in addition it had what was called a multiple cloning site or a poly cloning site that had a closely nested seq of RE cut sites and that was interesting but what was particularly interesting was that they ahd also gone thru all of the rest of the plasmid which was about two and a half thousand base pairs and made sure that none of these particular sites was available anywhere else in that plasmid. So instea d of cutting with a particular RE and having it cut here and here and here, it was just one single cut. So this was a very powerful tool because you could cut and be assured that you could get exactly what you were expectingvv
So RE have specific cut sites and the specific cut sites are incompatible between diff RE but you can imagine that if you had a fragment of dna that had an ecor1 stie and a hindiii site and you cut your plasmid with ecor1 and hindii , you would then be able to ligate a fragment into a vector, now when you do that what you're going to be doing is taking your gene of interest that you're going to study, you're going to cut it with the two enzymes, then you're going to mix the fragment in with the plasmid, allow base pairing to occur and then use the ligase to fit the whole thing back together and then you have the question of how it is that you're going to figure out, because all these htings are small and in solution and you can't tell them apart, you need to figureo ut how you tell whether you've done what you'd expect. Here the advantage is in that ?????? Lack z?? Bio synthesis gene that will turn the entire colony blue if you grow it in a particular substrate. Vvv There are two kinds of ecoli that won't grow. There's the regular ecoli, and there's the ecoli with the untransformed plasmid. The ones that don't pick up the color will be white. |
|
|
ethical questions related to plasmid modification etc?
|
- foreign dna and selection markers
- would you like to know if you have inherited the potential to have a disease in your 40s or 50s |
|
|
bioenergetics
|
how you acquire energy, how you get ATP energy from the food that you've consumed and how you use it for growth and development.
|
|
|
metabolism is
|
energy capture and it's energy use
|
|
|
thermodynamics is
|
the study of energy
|
|
|
high energy bonds are transduced into useable formats
|
NADP and ATP which are going to drive metabolism
|
|
|
What forms of energy are there?
|
Gravity, nuclear which holds nuclei together, light is important for photosynthesis, electrical/chemical is important for metabolism, mechanical is the ability to move things, heat is a source of energy at high rates and is a waste product which is always generated as a part of interaction.
|
|
|
first law
|
total amount of energy in a closed system is constant
|
|
|
the major forms of energy are present
|
in all sorts of compounds
|
|
forms of energy?
second law? |
gravitational, nuclear, light, electrical, chemical, mechanical, heat
some usable energy is lost when transferring from one type to another |
|
|
entropy?
|
The amount of unusable energy that is lost between the energy in the reactant molls and the product molls is called the disorder in the system and it's also called the entropy
|