Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
157 Cards in this Set
- Front
- Back
|
What metabolism reactions occur in the mitochondria?
|
B-oxidation, acetyl CoA synthesis, TCA cycle, oxidative phosphorylation.
|
|
|
What metabolism reactions occur in the Cytoplasm?
|
glycolysis, fatty acid synthesis, HMP shunt, protein synthesis (RER), steroid synthesis (SER)
|
|
|
What metabolism occurs in both cytoplasm and mitochondria?
|
Heme synthesis, Urea cycle, gluconeogenesis.
|
|
|
Enzyme terminology - what do each of these do?
1. Kinase 2. Phosphorylase 3. Phosphatase 4. Dehydrogenase 5. Carboxylase |
1. uses ATP to add high energy phosphate group onto a substrate.
2. adds an inorganic phosphate onto substrate without ATP 3. Removes a phosphate group from the substrate. 4. oxidises substrate. 5. Transfers CO2 groups with the help of biotin. |
|
|
What is the rate limiting Enzyme of this process?
Glycolysis |
Phosphofructokinase-1 (PFK-1)
|
|
|
What is the rate limiting Enzyme of this process?
Gluconeogenesis |
Fructose-1,6-bisphosphatase
|
|
|
What is the rate limiting Enzyme of this process?
TCA Cycle |
Isocitrate dehydrogenase
|
|
|
What is the rate limiting Enzyme of this process?
Glycogen synthesis |
Glycogen synthase
|
|
|
What is the rate limiting Enzyme of this process?
Glycogenolysis |
Glycogen phosphorylase
|
|
|
What is the rate limiting Enzyme of this process?
HMP Shunt |
Glucose-6-phosphate dehydrogenase (G6PD)
|
|
|
What is the rate limiting Enzyme of this process?
De Novo pyrimidine synthesis |
Carbamoyl phosphate sythetase II
|
|
|
What is the rate limiting Enzyme of this process?
De Novo purine synthesis |
Glutamine-PRPP amidotransferase
|
|
|
What is the rate limiting Enzyme of this process?
Urea cycle |
Carbamoyl phosphate synthetase I
|
|
|
What is the rate limiting Enzyme of this process?
Fatty acid Synthesis |
Acetyl-CoA carboxylase
|
|
|
What is the rate limiting Enzyme of this process?
Fatty acid Oxidation |
Carnitine acyltransferase I
|
|
|
What is the rate limiting Enzyme of this process?
Ketogenesis |
HMG CoA synthase
|
|
|
What is the rate limiting Enzyme of this process?
Cholestrol Synthesis |
HMG CoA Reductase
|
|
|
How are carbohydrates broken down starting at the mouth?
|
Starch (Glu-1,4-Glu chain), sucrose, lactose is broken down in the mouth randomly by saliva amylase. This process is continued by Pancreatic amylase in the SI until the starch is just dissaccarides such as maltose (Glu-1,4-Glu) and iso-maltose (Glu-1,6-Glu). These are then broken down into monosaccarides by brush border disaccharidases, to Glu, Gal, Fru which are taken up into the mucosal cells.
|
|
|
How is glucose moved into mucosal and renal cells?
|
Via a Na+/co-transporter.
|
|
|
How long does it take a plasma glucose level take to peak after a high Glu meal? At what time post-prandial is the Glu back to baseline levels?
|
45mins - peak
1hr 30mins - return to normal. |
|
|
What happens to Glu on entering cells?
|
It is phophorylated by Glucokinase (liver) or Hexokinase (most other cells).
|
|
|
How many ATP molecules are generated from Glu in aerobic metabolism?
|
32 via the malate-aspartate shuttle(heart/liver)
30 via glycerol-3-phosphate shuttle (muscle). 2 ATP anaerobically. |
|
|
How are the following transported?
1. Phosphoryl 2. Electrons 3. Acyl 4. CO2 5. 1-C units 6. CH3 groups 7. Aldehydes |
1. Phosphoryl (ATP)
2. Electrons (NADH, NADPH, FADH2) 3. Acyl (CoA, Lipoamide) 4. CO2 (biotin) 5. 1-C units (Tetrahydrofolate) 6. CH3 groups (SAM) 7. Aldehydes (TPP) |
|
|
What are the universal e- acceptors?
|
NAD+, NADP+, FAD+
|
|
|
In what processes are NAD+ used?
|
Catabolic, carrying reducing equivalents away.
|
|
|
In what processes are NADPH used?
|
Anabolic processes, respiratory bursts, P-450, glutathione reductase.
|
|
|
Describe the difference between Hexokinase and Glucokinase.
|
Hexokinase - ubiquitous, High affinity, low capacity (low Vmax), uninduced by insulin. Feedback = inhibited by Glu-6-P
Glucokinase - Liver and B-pancreatic cells, Low affinity (high Km), high capacity (high Vmax), induced by insulin. No direct feedback inhibition, phosphorylates Glu to sequester in liver (acts as a buffer). |
|
|
How does Fructose-2,6-Bisphosphate concentration regulate the glucose pathway?
|
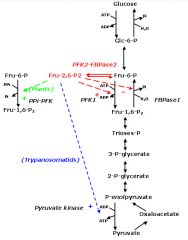
High concentrations upregulate PFK-1 activity converting more Fructose-6-P to Fructose-1,6-BP (the glycolysis pathway).
|
|
|
What affect does glucagon have on F-2,6-BP levels? What about Insulin?
|
increased glucagon - increased cAMP - increased pkA - increases fructose bisphosphatase 2 activity while decreasing phosphofructokinase 2 activity resulting in decreased F-2,6-BP. This results in a net decrease in glycolysis.
The presence of insulin does the opposite. Increasing glycolysis. |
|
|
What is the function of the pyruvate dehydrogenase complex?
|
converts pyruvate + NAD+ + CoA to acetyl-CoA + CO2 + NADH.
|
|
|
What 5 cofactors are required by the enzymes of pyruvate dehydrogenase complex?
|
Pyrophosphate (B1,thiamine, TPP)
FAD (B2) NAD (B3) CoA (B5) Lipoic acid. |
|
|
How does Arsenic inhibit pyruvate dehydrogenase complex? what symptoms are seen on Arsenic poisoning?
|
By inhibiting lipoic acid.
Symptoms - Vomiting, rice water stools, Garlic breath. |
|
|
What activates pyruvate dehydrogenase complex?
|
Exercise. increased NAD+/NADH ratio, ADP, Ca2+.
|
|
|
What occurs in pyruvate dehydrogenase deficiency? What are the findings? Treatment?
|
There are is a backlog of substrates (alanine, pyruvate) resulting in Lactic acidosis. Can be congenital or acquired.
Findings: neurologic defects Treatment: increased intake of ketogenic nutrients (high fat content or high lysine and leucine content) |
|
|
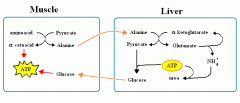
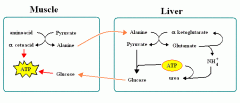
What are the ways pyruvate is metabolised?
|
The functions of different pyruvate metabolic pathways are -
1. ALAnine carries amino groups to the liver from the muscle. 2. Oxaloacetate can replenish TCA cycle or be used in gluconeogenesis. 3. Transition from glycolysis to the TCA cycle. 4. End of Anaerobic Glycolysis (major pathway to RBCs, leukocytes, kidney medulla, lens, testes, cornea). |
|
|
Describe the TCA cycle.
|
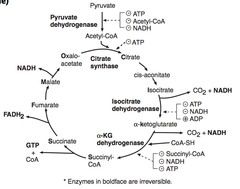
The TCA cycle produces 3 NADH, 1 FADH2, 2 CO2, 1 GTP per acetyl CoA. 2x for Glu.
a-Ketoglutarate dehydrogenasse requires the same co-factors as pyruvate dehydrogenase complex (B1,2,3,5, lipoic acid) The order of substrates through the cycle are as follows - Citrate, isocitrate, a-Ketoglutarate, Succinyl-CoA, Succinate, Fumarate, Malate, Oxalo-acetate. Mneumonic - Citrate Is Kreb's Starting Substrate For Making Oxalo-acetate. |
|
|
Describe the electron transport chain.
|
NADH and FADH2 donate e- to Complex I & II (respectively). As the e- travel along the complexes they generate a H+ gradient in the intermembranous space. This then diffuses down its gradient through ATPsynthase and generates ATP with the help of oxidative phosphorylation.
|
|
|
How many ATP are generated with NADH? FADH2?
|
NADH - 3ATP
FADH2 - 2 ATP. |
|
|
What is the mechanism of ETC inhibitors? Give examples.
|
Directly inhibit ETC, causing a decreased proton gradient and block ATP synthesis.
Rotenone, CN-, antimycin A, CO. |
|
|
How does ATPsynthase inhibitors work?
|
Directly in hibit mitochondrial ATPsynthase causing a increased H= Gradient. No ATP is generated because electron transport stops.
Oligomycin. |
|
|
How do uncoupling agents work? Give examples/
|
Increase membrane permeability, causing a decrease H+ gradient, and increasing O2 consumption. ATP synthesis stops but electron transport continues, produces heat
2,4-DNP, Aspirin, thermogenin in brown fat cells. |
|
|
What are the irreversible enzymes in Gluconeogenesis?
|
Pyruvate carboxylase
PEP carboxykinase Fructose-1,6-bisphosphatase Glucose-6-phosphatase |
|
|
Where does gluconeogenesis occur?
|
Primarily the liver, enzymes also in the kidney, intestinal epithelium.
|
|
|
How does odd-chain fatty acids produce Glu?
|
They yield 1 propionyl-CoA which can enter the TCA cycle as succinyl-CoA.
Even-chain FAs cannot produce new Glu, as they generate acetyl-CoA equivalents only. |
|
|
Describe the HMP shunt.
|
Provides NADPH from excess Glu-6-P, irreversible via Glu-6-P dehydrogenase; then converts ribulose-5-P reversibly via transketolases to Ribose-6-P. It also yields ribose for nucleotide synthesis and glycolytic intermediates. Occurs in the cytoplasm. No ATP is used or produced.
|
|
|
Where does the HMP shunt occur in the body?
|
Lactating mammary glands, liver, adrenal cortex, RBCs.
|
|
|
What is a respiratory burst? What are the steps involved?
|
Membrane bound NADPH oxidase converts O2 to radical oxygen species that are used in phagocytosis of microbes.
1. Membrane NADPH oxidase oxidises the O2 with an e-. 2. this free radical then binds to a H2O to form H2O2, via superoxide dismutase. 3. Cl- can be added to form HOCl- in the phagolysosome, which breaks down bacteria. |
|
|
What is deficient in those with chronic granulomatous disease?
|
NADPH oxidase. WBCs of pts with CGD can utilise H2O2 generated by invading organisms and convert it to ROIs. However, they are risk of infection by catalase +ve species (S.Aureus, Aspergillus) because they neutralise their own H2O2, leaving their WBCs w/o ROIs for fighting them.
|
|
|
What is the primary mechanistic fault in G6PD deficiency?
|
Unable to convert NADP+ to NADPH which is required to keep glutathione reduced. A deficiency results in a build up of free radicals and peroxides. This causes RBCs inparticular to be vunerable to oxidising agents resulting in Hemolytic Anaemia.hemolytic anaemia is often caused by drugs (sulphonamides, fava beans or primaquine) or infection.
|
|
|
What is the inheritance pattern of G6PD deficiency? Who is it more common in?
|
X-linked recessive; most common enzyme disorder; most common in african americans.
|
|
|
What is seen in a microscope with blood from a G6PD deficient pt?
|
Heinz bodies (oxidised Hb), Bite Cells (results from phagocytic removal of Heinz bodies).
|
|
|
Which disorder? What inheritance pattern?
involves a defect in Fructokinase. A benign, asymptomatic condition, since fructose does not enter cells. |
Essential Fructosuria. Autosomal recessive.
fructose appears in the blood and urine. Fructose metabolism is milder than analogous disorders of galactose. |
|
|
What disease results from a deficiency of aldose B? What is its inheritance pattern?
|
Fructose intolerance, Autosomal recessive.
|
|
|
What is the mechanism of the symptoms of fructose intolerance?
|
Symptoms: hypoglycemia, jaundice, cirrhosis, vomiting.
Fructose 1-P accumulates, causing a decrease in available phosphate. Results in inhibition of glycogenolysis, gluconeogenesis. treatment - reduce fructose and sucrose intake. |
|
|
Describe what is seen in galactokinase deficiency.
|
Galactitol accumulates if there is galactose in diet. relatively mild problems. Leads to infantile cataracts, can present with failure of familiar smile or tracking objects. Autosomal recessive.
|
|
|
Which disorder? What inheritance pattern?
galactose-1-phosphate uridyltransferase deficiency. Damage is caused by accumulation of toxic substances (galactitol, damages eyes) |
Galactosemia.
|
|
|
What are the Symptoms of galactosemia?
|
Failure to thrive, jaundice, hepatomegaly, infantile cataracts, mental retardation. More serious defects result in PO4 depletion.
treatment: limit galactose & lactose intake in diet. |
|
|
Describe sorbitol metabolism and how hyperglycemia results in sorbitol build up.
|
Glucose can be converted via aldose reductase to its alcohol counterpart sorbitol. This traps it within the cell. Some tissues then convert sorbitol to fructose by sorbitol dehydrogenase. In those tissues without sorbitol dehydrogenase, accumulation of sorbitol (causes osmotic damage) can occur in hyperglycaemia.
|
|
|
What symptoms occurs as a result of sorbitol accumulation?
|
Cataracts, retinopathy, peripheral neuropathy. (schwann cells, lens, retina, kidney cells only have aldose reductase)
|
|
|
Describe the enzyme deficiency, Symptoms and treatment of lactose deficiency.
|
Age-dependent or hereditary (african american, Asian) deficiency of teh brush border enzymes.May follow gastroenteritis.
Symptoms: Bloating, cramps, osmotic diarrhoea. Treatment: avoid dairy products, or add lactose pills to diet. |
|
|
Which form of amino acids are found in proteins?
|
L-form only.
|
|
|
What are the essential amino acids of the glucogenic, Glucogenic/ketogenic & ketogenic groups?
|
Glucogenic: Met, Val, Arg, His
Glucogenic/Ketogenic: Ile, Phe, Thr, Trp. Ketogenic: Leu, Lys. All need to be supplied by diet. |
|
|
What are the acidic and basic Amino acids?
|
Acidic: Asp, Glu (-ve charge at body pH)
Basic: Arg, Lys, His. n.b. Arg most basic, His has no charge at body pH. |
|
|
which 2 amino acids are in histones to bind DNA?
|
Lys and Arg.
|
|
|
What is the function of the Urea cycle?
|
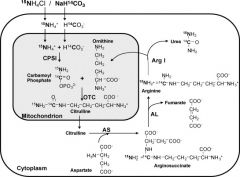
Amino acid catabolism results in the formation of common metabolites which serves as metabolic fuel. Excess Nh4+ is generated by this process. Urea is generated in the liver to be excreted by the kidney.
|
|
|
What is the order of the substrates in Urea formation?
|
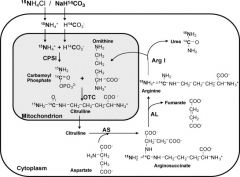
Ordinarily, Careless Crappers Are Also Frivolous About Urination.
Ornithine, Carbamoyl phosphate, Citrulline, Aspartate (donates NH4), Arginosuccinate, Fumarate & Arginine, Urea. |
|
Learn (or not)
|
That is all.
|
|
|
What is the Symptoms of Early onset (neo-natal) hyperammonemia?
|
Lethargy, irritability, poor feeding, Vomiting, Hyperventilation, grunting respiration, seizures.
|
|
|
What are the signs & symptoms of late onset hyperammonemia?
|
Intermittent Ataxia, intellectual impariment, Failure to thrive, Gait abnormality, Behaviour disturbances, Epilepsy, Recurrent Reye syndrome, protein avoidance, Rarely episodic headaches and cyclic vomiting.
|
|
|
What causes hyperammonemia?
|
USMLE: Can be acquired (liver disease) or hereditary (urea cycle enzyme deficiencies).
Medscape: A LLLLOOOONNNGGG LIST. including - Enzyme defects in urea cycle - NAGS, CPS I, OTC, AS, AL, arginase deficiencies. Organic acidemias. Congenital lactic acidosis - Pyruvate dehydrogenase, Pyruvate carboxylase, mitochondrial deficiencies. FA Oxidation defects. Di-basic amino acid transport defects Aphyxia Reye Syndrome Drugs (Valproate, topiramate, Carbamazepine, salicylate) Liver disease UTIs - urease +ve organisms Other causes - HSV neonatal pneumonitis, thyroid disease, hashimoto's encephalopathy, multiple myeloma (rare). |
|
|
How do you treat hyperammonemia?
|
limit protein in diet, Benzoate or phenylbutyrate (bind aa and lead to excretion), lactulose acidifies Gi and traps NH4+.
|
|
|
Which metabolic disorder? What is the inheritance pattern?
Most common urea cycle disorder. Interfers with the body's ability to eliminate ammonia. Often evident in first few days of life, but may present later in life. Excess carbamoyl phosphate converted to ortic acid. Signs & Symptoms - ortic acid in blood and urine, decreased BUN, symptoms of hyperammonemia. |
Orthine transcarbamoylase deficiency, X-linked recessive.
|
|
|
What is the amino acid derivatives?
Phenylalanine |
tyrosine to DOPA (& Thyroxine) to Dopamine (& melanin) to NE to Epinephrine.
|
|
|
What is the amino acid derivatives?
Tryptophan (2 pathways) |
1. with B6 - niacin to NAD+/NADP+
2. with BH4 - serotonin to melatonin |
|
|
What is the amino acid derivatives?
Histidine |
with B6 to Histamine.
|
|
|
What is the amino acid derivatives?
Glycine |
With B6 - porphyrin to heme.
|
|
|
What is the amino acid derivatives?
Arginine (3 pathways) |
Creatine
Urea Nitric oxide. |
|
|
What is the amino acid derivatives?
Glutamate (2 pathways) |
1. with B6 - GABA
2. Glutathione. |
|
|
What are the steps in catecholamine synthesis?
|
1. Phe to tyrosine (Phenoline hydroxylase, NADPH to NADP+, THB to DHB)
2. Tyrosine to dihydroxyphenylalanine, DOPA (tyrosine hydroxylase, NADPH to NADP+, THB to DHB) 3. DOPA to Dopamine (Dopa carboxylase, B6) 4. Dopamine to Norepinephrine (Dopamine B-hydroxylase, Vit C) 5. Norepinephrine to epinephrine (Phenylethanolamine N-methyltransferase, SAM) |
|
|
What are the breakdown components of -------- via MAO and COMT?
Dopamine NE Epi |
dopamine - HVA
NE - VMA Epi - metanephrine |
|
|
What is the mechanism of PKU?
|
Due to a decrease in phenylalanine hydroxylase or tetrahydrobiopterin co-factor. Tyrosine becomes essential, high phenylalanine in diet results in excess phenylketones in urine.
|
|
|
What is seen in a pt with phenylketouria?
|
Mental retardation, growth retardation, seizures, fair skin, eczema, musty body odor. Autosomal recessive. 1:10,000 incidence.
|
|
|
what is the treatment for PKU?
|
decrease phenylalanine in diet, increase tyrosine.
|
|
|
What is maternal PKU?
|
Lack of proper dietary therapy during pregnancy. Findings in the infant: microencephaly, mental retardation, growth retardation, congenital heart defects.
|
|
|
What is Alkaptouria?
|
Congenital deficiency of homogentistic acid oxidase in the degenerative pathway of tyrosine to fumurate. Autosomal recessive, benign disease.
|
|
|
What is seen in alkaptouria?
|
Dark connective tissue, brown pigmented sclera, urine turns black on prolonged exposure to air. may have debilitating arthralgias. Kidney stones in males are also common.
|
|
|
What disorder?
Congenital deficiency of either: Tyrosinase - autosomal recessive. Defective tysrosine transporters. |
Albinism, results from decreased conversion of tyrosine to melanin. Has variable inheritance - AR, X-linked recessive (ocular albinism)
|
|
|
What co-morbidity are people with albinism at risk for?
|
Skin cancer. lack melanin.
|
|
|
What are the 3 forms of homocystinuria? What are their inheritance patterns?
|
1. Cystathionine synthase deficiency (treatment: decrease Met, increase B12, folate & Cys in diet)
2. decreased affinity of cystathionine synthase for pyridoxal phosphate (treatment: increase B6 in diet) 3. Homocysteine methyltransferase deficiency All are autosomal recessive. |
|
|
What are the findings of homocystinuria?
|
increased homocysteine in urine, mental retardation, tall stature, osteoporosis, kyphosis, lens subluxation (down and inward), Atherosclerosis (stroke and MI).
|
|
|
What disorder?
hereditary defect of the renal tubular amino acid transporter for cysteine, ornithine, lysine & arginine in the PCT. Excess cysteine in the urine results in cysteine kidney stones. |
Cystinuria.
Autosomal recessive, common 1:7000. |
|
|
How do you treat Cystinuria?
|
Acetazolamide - alkalinizes the urine.
|
|
|
What disorder?
Severe CNS defects, mental retardation, death. Results from blocked degradation of Ile, Leu, Val due to decreased a-ketoacid dehydrogenase. Increases a-ketoacids in blood. |
Maple syrup urine disease.
Urine smells like maple syrup. |
|
|
What disorder?
autosomal recessive, defective neutral amino acid transporter on renal and intestinal epithelial cells. results in tryptophan excretion in urine and decresaed adsorption. leads to PELLAGRA. |
Hartnup disease.
|
|
|
How is glycogen regulated by insulin and glucagon/epinephrine?
|
Overall - Glycogen phophorylase kinase activates glycogen phosphorylase leading to glycogenolysis.
Activators 1. Ca2+/ calmodulin activates phosphorylase kinase so that glycogenolysis is co-ordinated with muscle activity. 2. Glucagon (acting at the liver) and epinephrine (liver & muscle) increase cAMP, via adenylyl cyclase, which activates in turn protein kinase A thereby activating Glycogen phosphorylase. Inhibitor Insulin works by dimerising receptor tyrosine kinase which in turn activates Protein phosphatase which inactivates glycogen phosphorylase & glycogen phosphorylase kinase. |
|
|
In glycogen what are the branching bonds?
|
a-(1,6) bonds
|
|
|
In glycogen what are the linking bonds?
|
a-(1,4) bonds.
|
|
|
What process does glycogen undergo in the skeletal muscle?
|
Glycogenolysis, forms Glu for use in exercise.
|
|
|
What process does glycogen undergo in Hepatocytes?
|
Glycogen is stored and undergoes glycogenolysis to maintain blood glucose.
|
|
|
Which glycogen storage disease? which enzyme is deficient?
Severe fasting hypoglycemia, increased glycogen in liverm increased blood lactate, hepatomegaly. |
Von Gierke's disease (type I), glucose-6-phosphatase
|
|
|
Which glycogen storage disease? which enzyme is deficient?
Cardiomegaly & liver and muscle involvement leading to early death. |
Pompe's disease (type II), Lysosomal a-1,4-glucosidase
|
|
|
Which glycogen storage disease? which enzyme is deficient?
Milder form of Type I, with normal blood lactate levels. Gluconeogenesis intact. |
Cori's disease (type III), Debranching enzyme (a-1,6-glucosidase)
|
|
|
Which glycogen storage disease? which enzyme is deficient?
increased glycogen in muscle, but cannot break it down, leading to painful muscle cramps. Myoglobinuria after exercise. |
McArdle's disease (Type IV), skeletal muscle glycogen phosphorylase.
|
|
|
Which lysosomal storage disease? Which enzyme deficiency? What substrate accumulates? Which inheritance pattern?
Peripheral neuropathy of feet/hands, angiokeratomas, cardiovascular/renal disease. |
Fabry's Disease.
Def enzyme: a-galactosidase A Substrate: Ceramide trihexoside Inheritance: X-linked recessive. |
|
|
Which lysosomal storage disease? Which enzyme deficiency? What substrate accumulates? Which inheritance pattern?
Hepatosplenomegaly, aseptic necrosis of the femur, bone crises, macrophages that look like crumpled tissue paper. |
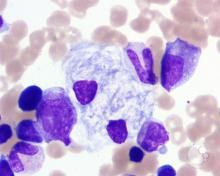
Gaucher's disease. Most common lysosomal enzymes.
Def enzyme: Glucocerebrosidase Substrate: Glucocerebroside Inheritance: autosomal recessive. |
|
|
Which lysosomal storage disease? Which enzyme deficiency? What substrate accumulates? Which inheritance pattern?
progressive neurodegeneration, hepatosplenomegaly, cherry-spot on macula, foam cells. |
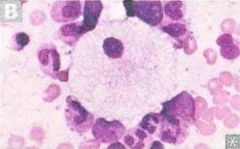
niemann-pick disease
Def enzyme: Sphingomyelinase Substrate: Sphingomyelin inheritance: Autosomal recessive |
|
|
Which lysosomal storage disease? Which enzyme deficiency? What substrate accumulates? Which inheritance pattern?
progressive neurodegeneration, developmental Delay, Cherry-spot on macula, lysosomes with onion skin appearance, NO HEPATOSPLENOMEGALY |
Tay-Sachs disease
Def enzyme: Hexosaminidase A Substrate: GM2 ganglioside Inheritance: Autosomal Recessive |
|
|
Which lysosomal storage disease? Which enzyme deficiency? What substrate accumulates? Which inheritance pattern?
Peripheral neuropathy, developmental delay, optic atrophy, globoid cells |
Krabbe's Disease
Def enzyme: B-galactocerebrosidase Substrate: Galactocerebrosidase Inheritance: Autosomal Recessive |
|
|
Which lysosomal storage disease? Which enzyme deficiency? What substrate accumulates? Which inheritance pattern?
Central and Peripheral demyelination with ataxia, dementia. |
Metachromic leukodystrophy
Def enzyme: Arylsulphatase A Substrate: Cerebroside sulfate Inheritance: Autosomal Recessive |
|
|
Which lysosomal storage disease? Which enzyme deficiency? What substrate accumulates? Which inheritance pattern?
Developmental delay, gargoylism, airway obstruction, corneal clouding, hepatosplenomegaly. |
Hurler's Syndrome (a Mucopolysaccaridose)
Def enzyme: a-L-iduronidase Substrate: Heparan sulphate, dermatan sulphate Inheritance: Autosomal Recessive |
|
|
Which lysosomal storage disease? Which enzyme deficiency? What substrate accumulates? Which inheritance pattern?
Mild Hurler's disease & aggresive behaviour, no corneal clouding. |
Hunter's syndrome (a mucopolysaccaridose)
Def enzyme: Iduronate sulphatase Substrate: Heparan Sulphate, Dermatan Sulphate Inheritance: X-linked recessive. |
|
|
Which 2 lysosomal storage diseases are x-linked recessive?
|
Hunter's syndrome & Fabry's disease
|
|
|
Which lysosomal diseases have and higher incidence in Ashkenazi jews?
|
Tay-Sachs, Niemann-Pick & some forms of Gaucher's disease.
|
|
|
What are the steps in Fatty acid synthesis?
|
Acetyl-CoA in the mitochondria matrix is transported to the cytoplasm via a citrate shuttle. In the cytoplasm it is converted to malonyl-CoA by the addition of a CO2 group from biotin. Malonyl-CoA is then converted to a FA (palmitate)
|
|
|
What are the steps in Fatty acid degradation?
|
FA & CoA are joined by FA-CoA synthetase to form Acyl-CoA which is then transported into the mitochondrial matrix by Carnitine shuttle. in the mitochondrial matrix, Acyl-CoA undergoes B-oxidation, converting it to acetyl-CoA groups.
Malonyl-CoA inhibits carnitine shuttle. |
|
|
What is seen in Carnitine deficiency?
|
results in an inability to transport LCFAs to the mitochondrial matrix. Findings include - Hypoketotic Hypoglycaemia, weakness, hypotonia.
|
|
|
What occurs in Acyl-CoA Dehydrogenase deficiency?
|
increased dicarboxylic acids, glucose, and ketones.
|
|
|
What ketone bodies are FA and amino acids converted to in the liver?
|
Acetacetate, B-hydroxybutyrate.
|
|
|
Which ketone body is not detected by urine test?
|
B-hydroxybutyrate
|
|
|
Describe the 3 processes in which ketone body production is increased. How do these processes increase ketone body production?
|
In prolonged starvation and DKA, oxaloacetate is depleted for gluconeogenesis.
In alcoholism, excess NADH shunts oxaloacetate to malate. Overall, TCA cycle is stalled resulting in shunting of glucose and FFA toward ketone body production. |
|
|
How are ketone bodies formed?
|
HMG-CoA breaksdown into 2 acetoacetate molecules. These can then be converted to B-hydroxybutyrate by NADH. Which is then excreted in the urine.
|
|
|
What are the energy sources used in seconds?
|
Stored ATP, Creatine phosphate.
|
|
|
What is the primary energy source while exercising for a few mins?
|
Anaerobic glycolysis
|
|
|
What is the primary energy source when you have been exercising longer than an hour?
|
Aerobic metabolism & FA oxidation.
|
|
|
What are the primary metabolic processes in the body when in a fed state?
|
Glycolysis and aerobic respiration. Isulin stimulates storage of lipids, proteins & glycogen.
|
|
|
What are the primary metabolic processes when fasting between meals.
|
Hepatic glycogenolysis. Also hepatic gluconeogenesis, adipose release of FFAs.
Glucagon and adrenaline stimulate fuel reserve use. |
|
|
What are the primary metabolic mechanisms in a Starvation state of 1-3 days?
|
Blood glucose level is maintained by-
1. hepatic glucogenolysis (reserves depleted in 1 day) 2. Adipose release of FFA 3. Muscle and liver shift from glu use to FFA use. 4. Hepatic gluconeogenesis from peripheral tissue lactate and alanine. and from adipose tissue glycerol & propionyl-CoA. |
|
|
What are the primary metabolic mechanisms in a starvation state of >3days?
|
Adipose stores - ketone bodies are the primary energy source for the brain and heart.
Once this source is depleted, vital protein degradation occurs resulting in organ failure and death. The amount of Adipose tissue determines the survival time. |
|
|
What is the rate limiting step in cholestrol formation?
|
HMG-CoA reductase. converts HMG-CoA to mevalonate. After formation 2/3 plasma cholestrol is esterified by LCAT.
|
|
|
How are lipids transported from the intestine to the liver?
|
In chylomicrons.
|
|
|
How are lipids transported from the liver to the adipose tissues and peripheral tissues?
|
Via VLDLs which are converted to IDLs then to LDLs which return to the liver.
|
|
|
What enzyme degrades TG in th eintestine?
|
pancreatic lipase
|
|
|
What degrades TG circulating in chylomicrons and VLDLs?
|
lipoprotein lipase.
|
|
|
What degrades TG in the IDLs?
|
hepatic TG lipase.
|
|
|
How is TG in adipocytes degraded into FFAs?
|
Hormone sensitive lipase.
|
|
|
How is cholestrol transporte in the body?
|
via HDLs/
|
|
|
What enzyme mediates transport of cholesterol to other lipoprotein particles?
|
Cholesterol ester transfer protein (CETP). Transfers cholesterol from HDL to VLDL, IDL, LDL.
|
|
|
What is the function of Apolipoprotein E?
|
mediates remnant uptake.
|
|
|
What is the function of Apolipoprotein A-1?
|
Activates LCAT.
|
|
|
What is the function of Apolipoprotein C-II?
|
Lipoprotein lipase cofactor.
|
|
|
What is the function of Apolipoprotein B-48?
|
mediates chylomicron secretion.
|
|
|
What is the function of Apolipoprotein B-100?
|
Binds to LDL receptors in the liver.
|
|
|
What apolipoproteins are present on chylomicrons?
|
E, A-1, C-2, B-48.
|
|
|
What apolipoproteins are present on Chylomicron remnants?
|
E, B-48.
|
|
|
What apolipoproteins are present on VLDL?
|
E, C-2, B-100.
|
|
|
What apolipoproteins are present on IDL?
|
E, B-100.
|
|
|
What apolipoproteins are present on LDL?
|
B-100
|
|
|
What apolipoproteins are present on HDL?
|
E, A-1.
|
|
|
What is the function of Chylomicrons?
|
Delivers TGs to the peripheral tissue.
Delivers cholestrol to liver in th eform of chylomicron remnants. Secreted by intestinal epithelial cells |
|
|
What is the function of VLDLs?
|
Delivers hepatic TGs to peripheral tissues.
Secreted by liver. |
|
|
Waht is the function of IDLs?
|
Formed in the degradation of VLDLs. Delivers Tgs and cholestrol to the liver. Where they are degraded to LDLs.
|
|
|
What is the functions of LDLs?
|
Delivers hepatic cholesterol to the periphery. Formed by lipoprotein lipase modification in the peripheral tissue. Taken up by receptor mediated endocytosis.
|
|
|
What is the function of HDLs?
|
mediates reverse cholesterol transport to the liver. Acts as a repository for apoC & apoE. Secreted by both the liver and intestine.
|
|
|
Which familial dyslipidemia? Describe the pathophysiology.
increased chylomicrons, elevated TGs & cholesterol. Pancreatitis, hepatosplenomegaly, eruptive/pruritic xanthomas. NO increased risk of atherosclerosis. |
Type I - hypercylomicronemia.
lipoprotein lipase deficiency or altered apoC-II. |
|
|
Which familial dyslipidemia? Describe the pathophysiology.
increased LDL, cholesterol. Accelerated atherosclerosis, tendon xantomas, corneal arcus. |
Type IIa - familial hypercholesterolemia.
Autosomal Dominant, absent or decreased LDL receptors. |
|
|
Which familial dyslipidemia? Describe the pathophysiology.
Increased VLDL & TGs pancreatitis |
type IV - hyper-triglyceridemia.
Hepatic overproduction of VLDL. |
|
|
Which familial dyslipidemia? Describe the pathophysiology.
Failure to thrive, steatorrhea, acanthocytosis, ataxia, night blindness. |
Abetalipoproteinemia.
Hereditary inability to synthesise lipoproteins deu to deficiency of ApoB-100 & B-48. Autosomal Recessive. symptoms appear in th efirst few months of life. intestinal biopsy shows accumulation in enterocytes due to inability to export lipid as chylomicrons. |