Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
199 Cards in this Set
- Front
- Back
|
pharmacokinetics
|
describes the absorption, distribution, metabolism, and exretion of inhaled or injected drugs (ie. what the body does to the drug)
|
|
|
pharmacodynamics
|
describes the responsiveness of receptors to drugs, and the mechanism by which these effects occur (ie. what the drug does to the body)
|
|
|
what are receptors?
|
the components of the cell that interact with the drug to initiate a sequence of events leading to pharamcologic effects
|
|
|
what determines selectivity of drug action?
|
receptors that recognize specific drugs
|
|
|
how does termination of a drugs effect occur?
|
with metabolism, excretion, or redistribution to inactive sites
|
|
|
what is the definition of a drug?
|
a chemical compound that produces pharmacologic effects as determined by its pharmacokinetics and pharmacodynamics
|
|
|
differing pharmacokinetics
|
the pharmacokinetic characteristics of drugs in healthy and ambulatory adults may differ from those with chronic diseases (esp. renal or hepatic dysfunction), and at extremes of age, hydration, nutrition, and skeletal muscle mass
|
|
|
what does the pharmacokinetic profile of a drug highly depend on?
|
the characteristics of the nonionized and ionized fraction of that drug
|
|
|
characteristics of the nonionized drug fraction
|
they tend to be pharmacologically active and lipid soluble
|
|
|
and the ionized fraction?
|
is inactive and water soluble
|
|
|
what is the ionization of a drug a function of?
|
the pK of the drug and the pH of the surrounding fluid
|
|
|
what happens when the pK and pH are identical?
|
50% of the drug exists in the ionized form
|
|
|
what happens with small changes in pH?
|
can result in large changes in the degree of ionization, esp if the pH and pK are similar
|
|
|
what is an example of an acidic drug?
|
barbiturates
|
|
|
and a basic drug?
|
opioids and local anesthetics
|
|
|
when are acidic drugs, such as barbiturates, highly ionized?
|
at an alkaline pH
|
|
|
and basic drugs, such as opioids and local anesthetics?
|
highly ionized at an acidic pH
|
|
|
what happens with ionized and nonionized drugs at lipid barriers?
|
nonionized drugs cross lipid barriers, such as renal tubules, GI tract, placenta, blood-brain barrier); ionized drugs dont cross
|
|
|
which is renally excreted, and which are metabolized hepatically?
|
ionized drugs tend to be renally excreted, and nonionized undergo hepatic metabolism
|
|
|
what else does lipid or water solubility determine?
|
absorption and elimination characteistics of drugs
|
|
|
definition of absorption
|
the rate at which a drug leaves its site of administration
|
|
|
membrane transport of drugs
|
occurs when drugs cross cell membranes by simple diffusion, by facilitated or carrier-mediated transport, and by active (energy-dependent) transport
|
|
|
simple diffusion
|
a bidirectional process in which the rate of transfer of a drug is proportional to the concentration gradient
|
|
|
what do highly lipid soluble and low-molecular weight drugs do at cell membranes?
|
diffuse passively across cell membranes
|
|
|
and large molecular weight drugs?
|
are transported across cell membranes by a carrier-mediated process, or an active transport system requiring energy
|
|
|
factors affecting drug absorption
|
particle size, lipid solubility, drug concentration, type of drug (crystalloid vs. colloid), area of absorption and local blood supply
|
|
|
absorption of tablets with a large particle size
|
may not disintegrate easily, and will have poorer absorption
|
|
|
absorption of drugs with high lipid solubility
|
will easily pass through membranes
|
|
|
absorption and concentration of drugs
|
a higher concentration of drug will favor its absorption
|
|
|
absorption of crystalloids vs. colloids
|
liquids or crystalloids are usually better absorbed, (ie. better solubility) than solids or colloids
|
|
|
absorption and local blood supply
|
a larger area of absorption and better local blood supply favor absorption
|
|
|
what do heat and vasodilation do to absorption, vs. shock and vasoconstriction?
|
heat and vasodilation increases absorption, whereas shock and constriction decrease it
|
|
|
ionization and absorption
|
the degree of ionization of a drug affects its absorption so that nonionized drugs, which are often lipid soluble, are absorbed rapidly, whereas ionized drugs tend to be water-soluble and are absorbed poorly
|
|
|
site of absorption of drugs
|
acidic drugs tend to be absorbed readily from the stomach, whereas basic drugs tend to be readily absorbed from the alkaline environment of the intestine
|
|
|
and finally, what is the last factor that determines absorption of drugs?
|
the route of administration
|
|
|
route of administration and absorption
|
IV administration of drugs allows for a predictable plasma concentration, whereas oral and IM injection of drugs often results in unpredictable absorption and is dependent of local blood flow
|
|
|
absorption of drugs from the GI tract
|
drugs absorbed from the GI tract, principally the small intestine, enter the portal venous blood, and pass through the liver before entering the systemic circulation and delivery to tissue receptors, known as first-pass hepatic metabolism
|
|
|
first-pass metabolism
|
for drugs that undergo extensive hepatic metabolism (eg. propranolol, lidocaine) this is the reason for such a large difference between effective oral and IV doses (ie. drug gets delivered to the receptors without first passing through the liver)
|
|
|
in addition to the liver, what other organ may play an important role in pharmacokinetics?
|
the lungs
|
|
|
pulmonary metabolism
|
the effect of the lungs on pharmacokinetics is reflected by basic lipophillic amines (eg. lidocaine, propranolol, and fentanyl) into lung tissue; the first-pass pulmonary effect may influence the peak arterial concentration of these drugs, and the lungs may subsequently serve as a reservoir to release drug back into systemic circulation
|
|
|
what does the passage of a drug through cell membranes depend on?
|
the pH of the drugs environment, the degree of ionization, the dissociation constant of the drug (pKa), protein binding, molecular weight, and lipid solubility
|
|
|
what is the pKa of a drug?
|
the pH at which the nonionized and ionized drug concentrations are equal
|
|
|
example of pKa and its effect on drug distribution
|
alfentanil has a more rapid onset of action than fentanyl because its pKa is close to physiologic pH, so most of the drug exists in the lipid-soluble, nonionized form at physiologic pH
|
|
|
what determines the concentration of a drug in the plasma and other tissues?
|
the binding of a drug to proteins
|
|
|
circulation of drugs after absorption
|
after absorption, drugs circulate in plasma in the unbound (free) form and in the form bound to protein
|
|
|
what proteins are drugs usually bound to?
|
acidic drugs to albumin, basic drugs to a1-acid glycoprotein
|
|
|
what does protein binding of drugs act to do?
|
act as a temporary reservoir of drug, and prevent large fluctuations of the unbound or free drug
|
|
|
what else can it do though?
|
protein binding can reduce a drugs metabolism and renal excretion by preventing its transport across renal tubular cell membranes
|
|
|
what things influence the binding of drugs to proteins?
|
increasing age, hepatic and renal disease, trauma, and surgery
|
|
|
what phenomenon can limit drug distribution and in whom does it occur?
|
ion trapping, in the fetus
|
|
|
ion trapping
|
fetal blood has a lower pH than normal adult blood, which means that drugs, such as local anesthetics and opioids which cross the placenta in the nonionized form, willl become ionized in the fetal ciculation, and because ionized drugs cant readily cross the placenta they accumulate in the fetal blood in a concentration gradient
|
|
|
what happens after systemic absorption of drugs?
|
the highly perfused tissues, eg. brain, heart, kidneys, and liver; recieve a proportionally larger amount of the total dose
for example, 75% of CO in a normal adult is delivered to the vessel-rich group of organs, which make up only 10% of body mass |
|
|
so due to this, what happens after IV administration of lipid-soluble drugs (eg. barbiturates, opioids)?
|
there will be rapid onset of CNS effects
|
|
|
redistribution
|
as the concentration of drugs in the plasma decreases below those in the highly-perfused tissues, drugs leave the highly-perfused tissues and are delivered to less well-perfused tissue beds, such as skeletal muscle and fat
|
|
|
thiopental redistribution
|
redistribution from the brain to inactive tissues is primarily whats responsible for awakening after a single dose of the drug; giving repeated doses can saturate inactive tissue sites decreasing the amount of sites available for redistribution, delaying awakening until metabolism can decrease plasma concentrations
|
|
|
fentanyl redistribution
|
the duration of action of fentanyl is normally short resulting from redistribution, but its effect can be prolonged when single, large doses or continuous infusions saturate inactive tissue sites available for redistribution
|
|
|
drug metabolism
|
done primarily in the liver, but to a lesser extent in the kidneys, lungs, and GI tract, its the conversion of pharmacologically active, lipid-soluble drugs to water-soluble, often inactive metabolites
|
|
|
what does increased water solubility do to a drug?
|
decreases the volume of distribution of that drug (Vd) and enhances its renal excretion
|
|
|
renal excretion of lipid-soluble drugs
|
they undergo minimal renal excretion because of the ease of which they are reabsorbed from the renal tubules into the pericapillary fluid
|
|
|
what else can metabolism do to a drug?
|
convert a prodrug into its active form, or cause toxic metabolites
|
|
|
what exactly metabolizes most drugs, and where are they located?
|
microsomal enzymes, and they are located principally in the hepatic smooth endoplasmic reticulum
|
|
|
where does the term microsomal enzymes come from?
|
from the fact that centrigugation of homogenized hepatocytes concentrates fragments of the smooth endoplasmic reticulum in whats called the microsomal fraction
|
|
|
what does the microsomal fraction contain?
|
the cytochrome P450 system, which likely contains many enzymes responsible for metabolism of many foreign compounds
|
|
|
what is enzyme induction?
|
stimulation of microsomal enzyme activity by drugs (classically phenobarbitol) which leads to accelerated metabolism of other drugs
|
|
|
what is the principle determinant of microsomal enzyme activity?
|
likely is genetic, which illustrates the large individual variations in the rate of metabolism of drugs among patients
|
|
|
what types of metabolic reactions can drugs undergo?
|
phase 1 or phase 2 reactions
|
|
|
phase 1 reactions
|
include oxidation, reduction, and hydrolysis
|
|
|
phase 2 reactions
|
includes conjugation
|
|
|
what does oxidation?
|
cytochrome p450 system
|
|
|
reduction?
|
by halogenated compounds
|
|
|
hydrolysis?
|
by procaine and amides
|
|
|
what do oxidation, reduction, and hydrolysis do to drugs?
|
they make them more water soluble by introducing polar groups like hydroxyl, amino, sulfhydryl, and carboxyl groups
|
|
|
conjugation
|
involves coupling a drug with an endogenous substrate, like glucuronate, acetate, or an amino acid so it can be excreted
|
|
|
how can drugs be excreted?
|
either unchanged or as metabolites
|
|
|
sites of drug excretion
|
primarily in the kidneys, but also the lungs, skin, bile, intestines, breast milk, saliva, and sweat
|
|
|
by what processes do the kidneys excrete drugs?
|
by passive glomerular filtration, active tubular secretion, and passive diffusion
|
|
|
nonionized drug excretion in the kidneys
|
they are easily filtered through the glomerulus, but they can also diffuse back across renal tubular cell membranes, leaving only a small amount of drug in the urine
|
|
|
ionized drug excretion
|
are not reabsorbed after filtration, and appear unchanged in the urine
|
|
|
how are many organic acids, like penicillin, excreted?
|
they are transported across renal tubules by systems that secrete naturally occuring substances, such as uric acid
|
|
|
passive diffusion of drugs
|
a bidirectional system where drugs diffuse across renal tubules according to a concentration gradient, lipid solubility, and pH
|
|
|
renal damage and excretion
|
renal damage impaires the ability of the kidneys to excrete drugs, which can result in high plasma concentrations and prologed duration of action of drugs
|
|
|
excretion and protein
|
protein binding can significantly alter the renal excretion of drugs, and hypoprotenemia can increase the amount of drug available for filtration
|
|
|
excretion and pH of urine
|
weak acids will be ionized in alkaline urine, and will therefore not be reabsorbed, while weak bases will be ionized in acidic urine and not be reabsorbed
|
|
|
how are volatile anesthetics excreted?
|
in the lungs
|
|
|
what happens to drugs excreted in the bile, and what are good examples of these?
|
they are reabsorbed repeatedly from the intestine (enterohepatic circulation), which plays a significant role in the excretion of drugs such as vecuronium and erythromycin
|
|
|
what are the pharmacokinetics of IV drugs influenced by?
|
the Vd of the drug and the clearance
|
|
|
what is the elimination half-time and what determines it?
|
its the rate at which the plasma concentration of a drug decreases with time, and it is also determined by Vd and clearance
|
|
|
what measures are more useful in characterizing the clinical response to a drug than elimination half-time?
|
context-sensitive half-time, and effect-site equilibrium
|
|
|
plot of drug concentration after an IV bolus of drug
|
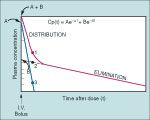
there will be 2 phases, the first is the distribution phase, the 2nd is the elimination phase
the distribution phase is the initial rapid decrease in plasma concentration, and corresponds to the distribution of drug from the circulation to the peripheral tissues the elimination phase is the gradual decrease in drug concentration and reflects its elimination from the central vascular compartment (ie. clearance) by the kidneys and liver |
|
|
distribution and elimination half-lives
|
are the time necessary to decrease the drug concentration 50% during the distribution or elimination phase
|
|
|
what is one of the most important pharmacokinetic variables to consider when setting a constant rate of IV drug infusion to maintain a steady-state plasma concentration?
|
clearance
|
|
|
clearance
|
the volume of plasma (ie. of the central compartment) cleared of drug in mL/min by renal or hepatic excretion, metabolism, or both
|
|
|
what happens when the rate of drug infusion exceeds clearance?
|
the plasma concentration of the drug will increase progressively, and cumulative drug effects occur
|
|
|
total clearance
|
is additive, and is the summation of the clearance rates for the liver, kidneys, and other routes
|
|
|
what are important factors determining clearance of a drug?
|
its concentration, and blood flow to the organ of clearance
|
|
|
what organ is most important for clearance of unchanged drugs or their metabolites?
|
the kidneys
|
|
|
clearance in the kidneys
|
water soluble compounds not bound to protein are the easiest to excrete, while protein-bound and lipid-spluble ones are harder, emphasizing how important metabolism is to convert lipid-soluble drugs to water-soluble metabolites
|
|
|
what are the most useful clinical indicators of the ability of the kidneys to eliminate drugs?
|
creatinine clearance, or serum creatinine concentration; the magnitude of increase of these will give a good estimate of the downward adjustment of drug dose required to prevent accumulation of drug in the plasma
|
|
|
volume of distribution (Vd)
|
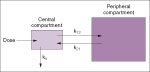
is a calculated number which reflects the apparent volumes of the compartments that constitute the compartmental model for that drug:
dose of drug administered IV/plasma concentration |
|
|
what factors will lead to a small Vd?
|
binding of drug to plasma proteins, a high degree of ionization, and low lipid solubility will all limit passage of drug to the tissues
|
|
|
examples of drugs with a small Vd
|
neuromuscular blockers
|
|
|
examples of drugs with a large Vd
|
thiopental and diazepam: they are nonionized lipid soluble drugs that readily pass into peripheral tissues from the circulation, and thus have low plasma concentrations
|
|
|
elimination half-time
|
the time necessary for the plasma concntration of drug to decrease 50% during the elimination phase
|
|
|
how many elimination half-times are required to completely eliminate a drug?
|
5
|
|
|
What will repeated doses of drugs equivalent to the initial dose at intervals more frequent than five elimination half times result in?
|
Cumulative drug affects
|
|
|
What will drug accumulation continue until?
|
The rate of drug elimination equals the rate of drug administered
|
|
|
What is the time necessary for drug to achieve steady-state plasma concentration (Cps) with intermittent doses?
|
Five elimination half-time
|
|
|
What is a common practice for achieving therapeutic concentration rapidly?
|
To administer a large initial IV dose i.e. loading dose, of drug to achieve a therapeutic concentration rapidly, and then give continuous or intermittent IV injections of decreased doses of drug to match the rate of elimination and maintain an optimal and unchanging plasma concentration
|
|
|
What must occur to the maintenance dose in the presence of renal or hepatic dysfunction?
|
It must be adjusted downward to prevent drug accumulation due to a prolonged elimination half-time
|
|
|
What is the descriptor most often used to characterize a drug's pharmacokinetic behavior?
|
Elimination half-time
|
|
|
However, what is the problem of elimination half-time?
|
It is useful only in the computation of central compartment drug concentration in a one compartment model, and it is of little value in describing the pharmacokinetics of drugs and multi-compartmental models
|
|
|
What is the anesthesiologist more interested in?
|
How long it will take the plasma concentration to decrease to a level that allows the patient to awaken, rather than the slope of the plasma drug concentration curve. Elimination half times alone provide virtually no insight into the rate of decrease in the plasma concentration after discontinuation of IV drug administration
|
|
|
So what then might the anesthesiologist wish to focus on?
|
Context-sensitive halftime
|
|
|
Context-sensitive halftime
|
The time necessary for the drug concentration to decrease to a predetermined percentage, for example 50%, 60%, 80%; after discontinuation of a continuous IV infusion of a specific duration; context refers to the duration of infusion
|
|
|
How are context-sensitive half times calculated?
|
Using computer simulation of multi-compartmental pharmacokinetic models drug disposition for drugs administered as continuous infusions during anesthesia
|
|
|
What is the context-sensitive halftime shown to be related to?
|
It increases in parallel with the duration of continuous IV administration, depending largely on the drugs lipid solubility and the efficiency of its clearing mechanisms: each drug will increase at a different rate based on the above factors
|
|
|
What is the relationship between the context-sensitive halftime and the drugs elimination half-time?
|
There is none
|
|
|
What does the time to recovery depend on?
|
How far the plasma concentration must decrease to reach levels compatible with awakening
|
|
|
Why is there a delay between the IV administration of a drug and the onset of its clinical effect?
|
This reflects the time necessary for the circulation to deliver the drug to its site of action, for example brain tissues. This delay reflects the fact that the plasma is not usually the site of drug action the circulation is merely the route by which the drug reaches its effect site
|
|
|
Effect site equilibrium time
|
The time for equilibrium between drug concentration in the plasma and the drug effect, for example time to produce a specific effect on the EEG
|
|
|
What drugs have short effect site equilibrium time?
|
remifentanil, alfentanil, thiopental, propofol
|
|
|
What drugs have a longer effect site equilibrium time?
|
fentanyl, sufentanil, midazolam
|
|
|
What is knowledge of the effect site equilibrium time important for?
|
Determining dosing intervals, especially when titrating IV drugs to a given clinical effect
|
|
|
What can failure to appreciate the importance of effect site equilibrium time result in?
|
Unnecessary or premature administration of drug
|
|
|
Bioavailability
|
The amount of active drug that is absorbed and reaches the systemic circulation; for example a drug that is absorbed from the intestine passes through the liver, first pass effect, before it reaches the circulation, and if the liver metabolizes the drug extensively the amount of active drug reaching the circulation and its site of action will be limited
|
|
|
What is an example of a drug degraded by the liver?
|
Propofol
|
|
|
What is the most important mechanism by which drugs exert pharmacologic effects?
|
By the interaction with a specific protein molecule in the lipid bilayer cell membranes, called a receptor
|
|
|
Examples of membrane receptors
|
Membrane or G protein coupled receptors, ligand gated ion channels, voltage sensitive ion channels, and enzymes
|
|
|
Drug interaction with receptors
|
A drug administered as an endogenous substance in contrast to endogenous hormones or neurotransmitters is an incidental passenger for these receptors; a drug – receptor interaction alters the function of a specific cellular component, which initiates or prevents a series of changes that characterize the pharmacologic effects of the drug
|
|
|
Receptor definition
|
Excitable transmembrane proteins that are responsible for transduction of biologic signals
|
|
|
Voltage sensitive ion channels
|
Depend on cell membrane voltage to open and close, for example sodium, chloride, potassium, calcium channels
|
|
|
Ligand gated ion channels
|
Function as receptor ion channel complexes in which the ion channel is an integral part of a larger and more complex transmembrane protein, for example nicotinic cholinergic receptors and amino acid receptors, e.g. GABA
|
|
|
What does activation of GABA – chloride channels, i.e. receptors, result in?
|
Cell hyperpolarization or an increase in ion conductance that prevents depolarization, thereby inhibiting neuronal activity; such activation enhances endogenous GABA mediated inhibition in the central nervous system, providing neurobiologic basis for the hypnotic and sedative effects of drugs such as benzodiazepines, barbiturates, and propofol
|
|
|
Prevalence of GABA receptors
|
Approximately 1/3 of all synapses in the CNS are responsive to GABA
|
|
|
What two parts contribute to the anesthetized state?
|
1. Sedation and loss of memory and consciousness – brain?
2. Analgesia and loss of pain sensation – spinal cord? |
|
|
How might opioids and alpha two agonists contribute to the anesthetized state?
|
Through inhibition of calcium channels and potassium channel activation – presynaptic inhibition
|
|
|
How do sedative – hypnotics, such as benzodiazepines propofol, etomidate, steroid anesthetics, contribute to the anesthetized state?
|
By activation of GABA a receptor chloride channels – leading to neuronal inhibition
|
|
|
How does ketamine work?
|
By inhibition of glutamate receptors
|
|
|
Guanine, G, proteins
|
Receptors that are important intermediaries in cell communication that reflect the molecular mechanisms of action of multiple classes of drugs, including opioids, sympathomimetic's, and anti-cholinergic's
|
|
|
G protein mechanism of action
|
An exogenously administered drug is recognized by its specific receptor, and the receptor – ligand interaction induces conformational changes, enabling the receptor to activate a specific gene protein which mediates the final cascade of biologic steps within the cell that leads ultimately to the pharmacologic and physiologic response characteristic of the administered drug
|
|
|
Examples of clinically important G protein coupled receptor systems
|
Include adrenergic, opioid, muscarinic, cholinergic, dopamine, and histamine receptors
|
|
|
Subtypes of G protein receptors
|
Multiple subtypes of receptors exist, including alpha 1 and two, beta one and two, muscarinic one and two, and histamine one and two receptors
|
|
|
Types of ion channels
|
Characterized as either voltage – sensitive or ligand – gated ion channels
|
|
|
Ligand – gated ion channels
|
Function as receptor – ion channel complexes in which the ion channel is an integral part of the larger and more complex transmembrane protein, for example glutamate activated-N-methyl-D-aspartate (NMDA) receptors, GABA receptors, nicotinic cholinergic receptors
|
|
|
What may be the principal molecular target for ketamine?
|
NMDA receptor
|
|
|
How do GABA – activated ion channels mediate the response to GABA?
|
By selectively allowing chloride ions to enter and thereby hyper polarizing neurons
|
|
|
Ion pumps
|
Are examples of excitable membrane proteins, such as the sodium/potassium – ATPase
|
|
|
Sodium potassium ATPase action
|
Action potentials activate sodium ion channels, allowing sodium the pass from outside to inside the cell. Sodium potassium ATPase then pump sodium out of the cell in exchange for potassium, returning the cell to its original cation composition
|
|
|
Digitalis mechanism of action
|
Inhibits the energy dependent sodium potassium ATPase ion pump, improving myocardial contractility
|
|
|
Agonist
|
A drug that initiates pharmacologic effects after combining with the receptor, therefore an agonist has a high efficacy and high affinity for the receptor; agonists induce stimulatory or inhibitory effects that mimic endogenous hormones and neurotransmitters
|
|
|
Antagonist drugs
|
Drugs that prevent receptor – mediated agonist effects by occupying agonist receptor sites, therefore they have the same affinity as the agonist for the receptor but their efficacy is poor
|
|
|
Mechanisms of action of antagonists
|
Can inhibit agonist effects by competitive inhibition, e.g. neuromuscular blocking drugs, or noncompetitive inhibition
|
|
|
What is a drug with an affinity equal to or less than that of the agonist but with lesser efficacy called?
|
A partial agonist
|
|
|
Inverse agonists, or super antagonists
|
Drugs that produce a response below the baseline response measured in the absence of the drug
|
|
|
Number of receptors
|
The number of receptors in the lipid cell membranes is dynamic, increasing or decreasing (up regulation or down regulation) in response to specific stimuli: for example prolonged beta adrenergic agonists as in the treatment of asthma are associated with tachyphylaxis and a concomitant decrease in the number of beta-adrenergic receptors, and conversely chronic beta blockade may result in increased number of beta-adrenergic receptors so an exaggerated response occurs if the block is abruptly reversed such as during the preoperative period
|
|
|
Receptors and aging
|
Changes in the responsiveness of receptors with no increase or decrease in the number of receptors may occur with aging, for example more isoproterenol is necessary to increase heart rate in the elderly compared with younger patients despite an unchanged number of receptors with aging
|
|
|
Dose – response curves
|
Depict the relationship between the dose of drug administered, or the resulting plasma concentration, and the resulting pharmacologic effect
|
|
|
How was the dose or concentration commonly graphed?
|
As the logarithmic transformation of dosage since this permits display a large range of doses
|
|
|
What causes the dose response curve to shift to the right?
|
A competitive antagonist or desensitization, or an agonist with a lower receptor affinity or potency
|
|
|
What causes a shift to the left?
|
An agonist with higher receptor affinity or potency
|
|
|
Drug potency on dose response curves
|
Is depicted by the location along the dose axis of the dose response curve
|
|
|
What is the dose required to produce a specific effect called?
|
Effective dose, ED, just the dose necessary to produce that effect in a given percentage of patients, for example ED 50, ED 90
|
|
|
What does increased affinity of a drug for its receptors due to the dose response curve?
|
Shifts it to the left
|
|
|
Drug potency in clinical practice
|
The potency of the drug makes little difference as long as the necessary dose of drug can be administered conveniently
|
|
|
Calculating drug dosage in common practice
|
Drug dosages commonly calculated on the basis of body weight, but when total body weight exceeds ideal body weight the total body weight will increasingly overestimate lean body mass, so in adults it is rarely necessary to scale dose to body weight more than 80 kg woman or 100 kg man
|
|
|
What influences the slope of the dose response curve?
|
The number of receptors that must be occupied before drug effect occurs
|
|
|
If the drug must occupy most receptors before its effect occurs what will happen to the slope of the dose response curve?
|
It will be steep
|
|
|
What are examples of drugs with steep dose response curves?
|
Neuromuscular blocking drugs and inhaled anesthetics
|
|
|
Clinically what does it mean when there is a steep slope of the dose response curve?
|
Small increases in dose evoke large increases in drug effect, for example one Mac of a volatile anesthetic prevents skeletal muscle movement in response to a surgical skin incision in 50% of patients, ED 50, where is a further modest increase to 1.3 Mac prevents movement and at least 95%, ED 95, of patients
|
|
|
What is also true when the dose response curve is steep?
|
The difference between a therapeutic and toxic concentration may be small, which is true for volatile anesthetics where there is a small difference between doses that produce desirable central nervous system depression and undesirable cardiopulmonary depression
|
|
|
Drug efficacy
|
The maximal effect of a drug as depicted by a plateau in the dose response curve
|
|
|
What may limit dosage to below the concentration associated with maximal effect?
|
Undesirable side effects of the drug
|
|
|
What is the ceiling effect?
|
A phenomenon where the degree of effect produced by increasing doses of drug eventually reaches a steady level, and if the dose of drug exceeds the ceiling dose there is no further increase in therapeutic effect and undesirable side effects may predominate
|
|
|
Ceiling dose
|
The dose at which the ceiling effect is obtained
|
|
|
Are efficacy and potency related?
|
Not necessarily
|
|
|
Individual responses to drugs
|
Individual responses may very as reflections of differences and pharmacokinetics, e.g. renal liver or cardiac function or patient's age, or pharmacodynamics, such as enzyme activity in genetic differences. For example benzodiazepines have a more pronounced effect in the elderly than in young adults
|
|
|
Malignant hyperthermia
|
A hyper metabolic state triggered in some individuals in response to administration of succinylcholine or volatile anesthetics, which represents a genetic pharmacodynamic abnormality
|
|
|
Therapeutic index
|
The ratio between the lethal dose in 50% of patients, LD50, and the effective dose in 50% of patients, ED50
|
|
|
Therapeutic index and safety
|
The higher the therapeutic index of a drug, the safer it is for clinical administration because the LD is far above the ED
|
|
|
Additive effect
|
When the pharmacologic effect of two or more drugs administered together is equivalent to the summation of their individual effect
|
|
|
Synergistic response
|
When the pharmacologic effect of two or more drugs administered together is greater than the sum of the individual effects
|
|
|
Time synergism
|
The prolongation of action of one of the drugs when two drugs are administered together, for example combination of lidocaine and epinephrine increases the duration of action of lidocaine
|
|
|
Competitive antagonism
|
The competitive antagonist can usually be displaced from the receptor by administration of high doses of agonist, it shifts the dose response curve to the right, and it is usually reversible
|
|
|
Noncompetitive antagonism
|
When bounde to receptors noncompetitive antagonists produce a confirmational change in the receptor which results in diminished receptor response when exposed to agonist even at high doses, the dose response curve shifts to the right, the slope is reduced and the maximum pharmacologic response diminishes
|
|
|
Is noncompetitive antagonism reversible or irreversible?
|
It can be either
|
|
|
Drug stereospecificity
|
The molecular interactions which are the foundation of pharmacokinetics and pharmacodynamics are stereoselective or stereospecific, emphasizing the drugs are expected to interact with other biological components, e.g. receptors, in a geometrically specific way.
|
|
|
Racemic mixture
|
When two isomers, dextro (D) and levo (L), of opposite shape are present in equal proportions
|
|
|
Administration of the racemic mixture of drugs
|
May represent pharmacologically two different drugs with distinct pharmacokinetic and pharmacodynamic properties, the two isomers may have different rates of absorption, metabolism, and excretion and different affinities for receptor binding sites. And although only one isomer is active it is possible that the other isomer contributes to its side effects
|
|
|
Racemic mixture examples
|
D – bupivacaine remains in sodium ion channels for a longer period than the L – isomer which may result in cardio toxic effects; but ropivacaine and levobupivacaine are present only has the L – isomer and are not as likely to produce cardio toxic effects as bupivacaine
D – ketamine is predominantly hypnotic and analgesic, whereas L – ketamine is the likely source of the drugs unwanted side effects |
|
|
How should the inactive isomer in a racemic mixture be regarded?
|
As an impurity
|
|
|
Drug tolerance
|
When a large dose of drug is required to elicit an effect that is usually produced by smaller therapeutic doses of the drug; it can be natural or acquired
|
|
|
Natural tolerance
|
Can be species or racial specific
|
|
|
Acquired tolerance
|
Developed in response to repeated administration of the drug and can be either tissue tolerance or cross – tolerance
|
|
|
Tissue tolerance
|
Is tolerance confined to certain pharmacologic effects such as tolerance to euphoric effects of morphine but not its constipating effects
|
|
|
Cross tolerance
|
Occurs when an individual develops tolerance to a specific drug, for example alcohol, and other drugs, e.g. sedative – hypnotics, inhaled anesthetics, producing similar pharmacologic effects
|
|
|
Tachyphylaxis
|
Acute tolerance to the pharmacologic effects of certain drugs, for example ephedrine and amphetamines, which may occur when they administered at short intervals. The mechanism responsible is unclear but may reflect depletion of norepinephrine stores or altered dissociation of drug from its receptor sites
|
|
|
Drug dependence
|
The psychic or physical state characterized by behavioral responses including a compulsion to take the drug on a continuous or periodic basis to experience its effects and sometimes to avoid the discomfort of its absence; tolerance may or may not occur, and a withdrawal syndrome which may be life-threatening may develop on discontinuation of drug
|
|
|
Drug interactions
|
Simultaneously administered drugs can alter each other's pharmacokinetic and pharmacodynamic behaviors, for example ranitidine or metoclopramide can alter drug absorption by changing the pH of gastric secretions and G.I. motility
|
|
|
Cholinesterase inhibitors
|
Antagonize neuromuscular block by increasing the amount of acetylcholine, which displaces non-depolarizing neuromuscular blocking drugs from nicotinic receptors
|