![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
205 Cards in this Set
- Front
- Back
|
Anemias - Parte 1 Resumão |

|
|
|
Hematopoiese |

|
|
|
Vida média das hemácias |
120 dias |
|
|
Vida média das plaquetas |
7-10 dias |
|
|
Vida média dos granulócitos |
6-8 horas |
|
|
Vida média dos linfócitos |
Muitos anos |
|
|
Principais mediadores da hematopoiese (2) |
Interleucinas e fatores de crescimento
Ex: eritropoietina e GM-CSF |
|
|
Local de eritropoiese nas primeiras semanas de vida embrionária |
Saco vitelínico |
|
|
Local de eritropoiese durante o 2° trimestre da gestação |
Fígado, baço e linfonodos |
|
|
Local de eritropoiese nos estágios finais de vida fetal |
Medula óssea passa a ser o principal terreno da hematopoiese |
|
|
Local de eritropoiese após o nascimento |
Exclusivamente na medula óssea |
|
|
Em vigência de determinadas patologias, o que acontece em relação à produção de células hematológicas? |
Volta a ocorrer nos locais extramedulares (linfonodos, fígado e baço) |
|
|
Primeira célula no interior da medula óssea identificada como pertencente à série eritroide |
Pró-eritroblasto |
|
|
Onde ocorre a síntese de hemoglobina? |
Na mitocôndria das células da linhagem vermelha (de pró-eritroblasto até reticulócito) |
|
|
Composição da hemoglobina |
4 cadeias de globinas + 4 porções heme |
|
|
Composição do heme |
4 anéis aromáticos (protoporfirina) + um átomo de íon ferroso (Fe+2), capaz de ligar o O2 |
|
|
Tipos de hemoglobinas no adulto normal (3) |
Hb A (97%): 2 cadeias alfa + 2 cadeias beta Hb A2 (2%): 2 cadeias alfa + 2 cadeias delta Hb F (1%): 2 cadeias alfa + 2 cadeias gama |
|
|
Doenças genéticas caracterizadas pela redução ou perda da produção de uma determinada cadeia de globina |
Talassemias |
|
|
Desordens (hereditárias ou adquiridas) caracterizadas por um defeito na síntese do heme |
Anemias sideroblásticas |
|
|
O que são sideroblastos em anel? |
Eritroblastos com depósito de ferro mitocondrial (em volta do núcleo) |
|
|
Valores normais da série vermelha (Hb e Ht - mulheres e homens) |

|
|
|
Manifestações clínicas da síndrome anêmica |

|
|
|
No geral, os sintomas do quadro anêmico provém do que? |
Do prejuízo na capacidade carreadora de O2 do sangue, predispondo à hipóxia tecidual e estimulando o coração a aumentar o débito cardíaco de forma compensatória |
|
|
2 principais mecanismos que amenizam a hipóxia tecidual decorrente da anemia |
1) Aumento do débito cardíaco 2) Aumento do 2,3 DPG (glicose difosfato) na hemácia - este metabólito reduz a afinidade da Hb pelo O2 |
|
|
Investigação etiológica das anemias
O que procurar na anamnese? (2) |
1) Tempo de instalação dos sintomas 2) Sintomas associados |
|
|
Investigação etiológica das anemias O que procurar no exame físico? (5) |
1) Glossite e queilite angular - anemias carenciais 2) Icterícia - anemias megaloblástica e hemolítica 3) Esplenomegalia significativa - anemia hemolítica, hiperesplenismo e neoplasias hematológicas 4) Petéquias - anemia aplásica e leucemias agudas 5) Deformidades ósseas na face e crânio - talassemia |
|
|
Exames para o diagnóstico etiológico da anemia |

|
|
|
O que é VCM? |
Volume Corpuscular Médio
Volume médio das hemáceas VCM = Ht × hematimetria
80-100 fL |
|
|
O que é HCM? |
Hemoglobina Corpuscular Média
Massa de hemoglobina média das hemácias HCM = Hb × hematimetria
28-32 pg |
|
|
O que é CHCM? |
Concentração de Hemoglobina Corpuscular Média CHCM = HCM/VCM × 100
32-35 g/dL |
|
|
O que é RDW? |
Índice de anisocitose Indica a variação de tamanho entre as hemácias 10-14% Obs.: útil para diferenciar talassemia minor e anemia ferropriva! |
|
|
A presença de pancitopenia sugere...? |
Anemia aplásica Leucemia aguda |
|
|
A presença de bicitopenia (síndrome de Evans) surge...? |
Anemia autoimune |
|
|
Anemia micro e hipo com trombocitose sugere? |
Anemia ferropriva |
|
|
Anemia + leucocitose + trombocitose sugere...? |
Sangramento agudo Síndromes mieloproliferativas |
|
|
A presença de reticulocitose indica quais grupos de anemia? (2) |
Anemias hemolíticas Anemia por hemorragia aguda Mielograma: hiperplasia eritroide |
|
|
Contagem diferencial dos leucócitos |

|
|
|
Anemias microcíticas e hipocrômicas (5) |
Anemia ferropriva Talassemia Anemia de doença crônica de longa duração Anemia sideroblástica, forma hereditária Anemia do hipertireoidismo |
|
|
Anemias macrocíticas (9) |
Anemia megaloblástica Síndromes mielodisplásicas, incluindo a forma adquirida da anemia sideroblástica Anemia aplásica Etilismo Drogas do tipo AZT e MTX Anemia da hepatopatia crônica Anemia do hipotireoidismo Anemais hemolíticas (exceto talassemias) Anemia da hemorragia aguda |
|
|
Anemias normocíticas e normocrômicas (11) |
Anemia ferropriva - fase inicial Anemia de doença crônica Anemia da IRC Anemia da hepatopatia crônica Anemia das endocrinopatias - hipotireoidismo, hipoadrenalismo Anemia aplásica Mielodisplasias Ocupação medular Anemias hemolíticas Anemia por sangramento agudo Anemia multicarencial - ferropriva e megaloblástica |
|
|
Anemias hipoproliferativas/sem reticulocitose |
Carenciais, distúrbios medulares |
|
|
Anemias hiperproliferativas Com reticulocitose |
Hemolítica ou sangramento agudo |
|
|
Correções do percentual reticulocitário para o grau de anemia (2) |
1) Índice de Reticulócitos Corrigido (IRC) = Ht/40 × %retic ou Hb/15 × %retic 2) Índice de Produção Reticulocitária (IPR) = Ht/40 × %retic÷2 ou Hb/15 × %retic÷2 |
|
|
Anemia que cursa com hiperbilirrubinemia indireta, aumento do LDH e redução da haptoglobina |
Anemia hemolítica |
|
|
Confirmam o diagnóstico de anemia ferropriva |
Saturação de transferrina < 15% --> Cálculo: Ferro÷TIBC (capacidade de ligação total da transferrina) |
|
|
Corpúsculos de Howell-Jolly Situações em que aparece |
Hipoesplenismo! Esplenectomia Anemia falciforme Doença celíaca (15%) |
|
|
Corpúsculos de Pappenheimer Situações em que aparece |
Anemia sideroblástica Hipoesplenismo |
|
|
Corpúsculos de Heinz Situações em que aparece |
Hemoglobinopatias Deficiência de G6PD |
|
|
Diagnósticos que só são possíveis com o mielograma (5) |
Anemia aplásica Leucemia Mielodisplasia Mieloma múltiplo Anemia sideroblástica |
|
|
Compartimentos armazenadores de ferro (2) |
Ferritina Hemossiderina |
|
|
Proteínas capazes de ligar o ferro (3) |
Ferritina Hemossiderina Transferrina |
|
|
Principal responsável pelo armazenamento de ferro no organismo |
Ferritina 20-200 ng/ml 1 ng/ml = 10 mg de ferro armazenado |
|
|
Proteína responsável pelo transporte de ferro no plasma |
Transferrina |
|
|
Tipos de ferro alimentar (2) |
Ferro heme: origem animal Ferro não heme: origem vegetal |
|
|
Tipo de ferro alimentar melhor absorvido e que não sofre nenhuma influência de outras substâncias alimentares |
Ferro heme |
|
|
Estimulantes e inibidores da absorção de ferro não heme |

|
|
|
Estimula a absorção intestinal de ferro não heme e pode figurar como adjuvante na prescrição de reposição oral deste metal |
Vitamina C |
|
|
É um importante compartimento armazenador de ferro |
Mucosa intestinal |
|
|
É a principal via de eliminação do ferro e de proteção contra uma sobrecarga dietética |
Descamação das células da mucosa intestinal (via fecal) |
|
|
Função da hepcidina |
Ela tem a capacidade de se ligar à ferroportina, inibindo o transporte de ferro pela membrana basolateral do enterócito em direção ao plasma |
|
|
O que inibe e o que estimula a síntese de hepcidina? |
Hipóxia inibe Ferro e citocinas inflamatórias estimulam |
|
|
O organismo humano não é capaz de eliminar ativamente o ferro já estocado em seu interior. V ou F? |
V |
|
|
3 doenças que aumentam patologicamente a absorção intestinal de ferro |
Talassemias Anemia sideroblástica Hemocromatose primária (hereditária) |
|
|
Principal causa de anemia ferropriva |
Sangramento CRÔNICO |
|
|
1 ml de sangue contém quantos mg de ferro? |
1 ml de sangue = 0,5 mg de ferro |
|
|
Qual sangramento nem sempre causa anemia ferropriva? |
Sangramento AGUDO! |
|
|
TIBC = ? |
Capacidade de ligação total de ferro |
|
|
A saturação de transferrina sempre aumenta com o aumento do ferro sérico. V ou F? |
F Isso nem sempre acontece pois a se a massa total de transferrina também aumentar (ex: o fígado produziu mais transferrina), a saturação não sofrerá variações |
|
|
O que representa o TIBC? |
O somatório de todos os sítios de ligação de todas as moléculas de transferrina circulantes, ou seja, o somatório dos sítios ligados com os sítios livres |
|
|
Como calcular o TIBC? |
TIBC = (sítios de transferrina ligados ao ferro) + (sítios de transferrina não ligados ao ferro) Ou seja: TIBC = (Ferro sérico) + (Capacidade Latente de Fixação de Ferro) |
|
|
Valores normais: Fe serico e TIBC |
Fe sérico: 60-150 mcg/dl TIBC: 250-360 mcg/dl |
|
|
Cálculo da saturação da transferrina |

Sat transferrina = 30-40% |
|
|
Causa mais comum de anemia em crianças |
Anemia ferropriva |
|
|
Sinais e sintomas da ferropenia |
Glossite (ardência e língua despapilada) Queilite angular (lesão nos ângulos da boca) Unhas quebradiças e coiloníquia (unhas em colher) Esplenomegalia de pequena monta Perversão do apetite (pica ou parorexia) - desejo de comer alimentos de baixo valor nutricional Síndrome de Plummer-Vinson ou Paterson-Kelly (disfagia) Fraqueza muscular desproporcional ao grau de anemia |
|
|
Sintomas mais comuns de ferropenia em crianças (2) |
Anorexia e irritabilidade |
|
|
Hemograma na anemia ferropriva |
Fase inical: anemia normo/normo Segunda fase: anemia micro/hipo
Média: Hb: 8 VCM: 74 CHCM: 28 RDW aumentado (16%)
Trombocitose Leucometria normal
|
|
|
Anemia ferropriva frequentemente se associa à qual anemia? |
Megaloblástica (anemia multicarencial)
Nesses casos, o VCM pode estar desde diminuído até aumentado |
|
|
Diante de uma anemia com acentuada microcitose (VCM < 72), pensar em quais tipos de anemia? (2) |
Ferropriva - RDW aumentado Talassemia - RDW normal |
|
|
Reticulócitos na anemia ferropriva |
Normal |
|
|
Laboratório do ferro na anemia ferropriva |
Ferro sérico baixo (< 30 mg/dl) Ferritina sérica baixa (< 15 ng/ml) Transferrina sérica aumentada TIBC elevado (> 400 mg/dl) Saturação da transferrina diminuída (< 15%) |
|
|
Primeiro parâmetro a se alterar na anemia ferropriva |
Ferritina sérica |
|
|
Hematoscopia na anemia ferropriva |
Fase inicial: anisocitose Anemia grave: poiquilocitose Anisopoiquilocitose! |
|
|
A análise do ferro medular pode permitir a diferenciação entre anemia ferropriva e anemia de doença crônica quando os níveis de ferritina forem inconclusivos. V ou F? |
V |
|
|
Estágios na deficiência de ferro (3) |
1) Depleção dos estoques de ferro 2) Eritropoiese deficiente em ferro 3) Anemia ferropriva |
|
|
Qual é o exame "padrão-ouro" para o diagnóstico de anemia ferropriva? Qual achado confirma seu diagnóstico? |
Aspirado/biópsia de medula óssea e coloração para ferro com o corante azul da Prússia Ausência de ferro nos macrófagos e eritroblastos da medula |
|
|
Dose de ferro elementar recomendada para a correção da anemia ferropriva |
60 mg de Fe elementar (=300 mg de sulfato ferroso) 3-4x ao dia Em crianças: 5 mg/kg/dia |
|
|
Quando e como tomar o sulfato ferroso? |
A absorção do sulfato ferroso é melhor quando administrado de estômago vazio, 1-2h antes das refeições, de preferência associado à vitamina C ou ao suco de laranja |
|
|
Como saber se o tratamento da anemia ferropriva está sendo eficaz? |
Avaliando a contagem de reticulócitos
Pico: 5-10 dias |
|
|
Qual a duração do tratamento da anemia ferropriva? |
A reposição de ferro deve durar 3-6 meses após a normalização do hematócrito (total de 6-12 meses), no intuito de reabastecer os estoques corporais desse elemento |
|
|
Como fazer o controle do tratamento? |
Com a ferritina sérica, que deve chegar a valores acima de 50 ng/ml |
|
|
Indicações de ferro parenteral (4) |
1) Síndromes de má absorção do duodenojejunais, como a doença celíaca 2) Intolerância às preparações orais 3) Anemia ferropriva refratária a terapia oral, apesar da adesão terapêutica 4) Necessidade de reposição imediata dos estoques de ferro |
|
|
Anemia de doença crônica Definição |
Estado anêmico moderado que frequentemente se desenvolve nos pacientes que apresentam condições inflamatórias ou neoplásicas, sempre quando estas se fazem presentes por um período superior a 20-60 dias |
|
|
Todas as doenças crônicas justificam o desenvolvimento de ADC. V ou F? |
F Em geral são aquelas doenças que, de alguma forma, aumentam as citocinas pró-inflamatórias |
|
|
Exemplos de condições que originam a ADC |

|
|
|
Como a hepcidina é produzida? |

|
|
|
Laboratório na ADC |
1) Ferro sérico baixo 2) Ferritina sérica normal ou alta 3) Saturação de transferrina levemente baixa 4) TIBC normal ou baixo |
|
|
Quando suspeitar de anemia ferropriva sobreposta em pacientes com ADC ou doença inflamatória/infecciosa/neoplásica? |
Quando ferritina for inferior a 30 ng/ml |
|
|
Qual é a principal causa de anemia micro/hipo? |
Anemia ferropriva |
|
|
Qual é a principal causa de anemia normo/normo? |
Anemia ferropriva |
|
|
Qual é a segunda principal causa de anemia micro/hipo? |
ADC* *Se VCM < 72, a segunda causa é talassemia |
|
|
Qual é a segunda principal causa de anemia normo/normo? |
ADC |
|
|
Qual é a principal causa de anemia em pctes hospitalizados? |
ADC |
|
|
Por que na anemia megaloblástica temos hemácias aumentadas de tamanho? |
Na anemia megaloblástica temos uma alteração da síntese do DNA. Com isso, o núcleo da célula amadurece de forma lenta, e a divisão celular se dá mais tardiamente. Entretanto, o citoplasma não sofre alterações à espera da divisão celular, que não vem... |
|
|
Por que na anemia megaloblástica temos hemácias aumentadas de tamanho? |
Na anemia megaloblástica temos uma alteração da síntese do DNA. Com isso, o núcleo da célula amadurece de forma lenta, e a divisão celular se dá mais tardiamente. Entretanto, o citoplasma não sofre alterações à espera da divisão celular, que não vem... |
|
|
O que quer dizer exatamente megaloblastose? |
É uma alteração no núcleo dos precursores eritroides (que só pode ser observada na medula óssea) |
|
|
Quando pensamos em anemia megaloblástica, quais são as principais etiologias? |
Deficiência de vitamina B12 (cobalamina) e ácido fólico |
|
|
Qual é a alteração no sangue periférico que mais sugere anemia megaloblástica? |
Além da macrocitose, existe outra alteração clássica: a hiperpigmentação do núcleo dos neutrófilos |
|
|
Qual é a alteração no sangue periférico que mais sugere anemia megaloblástica? |
Além da macrocitose, existe outra alteração clássica: a hiperpigmentação do núcleo dos neutrófilos |
|
|
Qual é o grupo que tem a maior chance de desenvolver anemia perniciosa? |
Mulheres brancas de meia-idade |
|
|
Qual é o mecanismo patogênico da anemia perniciosa? |
É uma condição autoimune - destruição e atrofia da mucosa do estômago, levando à redução da produção de FI |
|
|
Quais os principais autoanticorpos na anemia perniciosa? |
Anticélula parietal: 90% dos casos Anti-FI: 60% dos casos |
|
|
Existe associação da anemia perniciosa com alguma doença neoplásica? |
Sim Com adenocarcinoma gástrico |
|
|
Existe associação da anemia perniciosa com outras doenças autoimunes? |
Sim, com a doença de Graves, Hashimoto, vitiligo, insuficiência adrenal idiopática e hipoparatireoidismo |
|
|
Existem outras causas de macrocitose que não a anemia megaloblástica? |
Sim. As principais são: mielodisplasias, anemia aplástica, anemia sideroblástica forma secundária, anemia da hepatopatia, alcoolismo, hipotireoidismo, anemia hemolítica grave e toxicidade de algumas medicações |
|
|
Quando devemos pensar em anemia megaloblástica? |
Em toda anemia macrocítica, principalmente aquelas que cursam com o VCM muito aumentado (VCM > 120 fL é virtualmente patognomônico de megaloblastose) |
|
|
Qual é o achado no sangue periférico considerado quase patognomônico de anemia megaloblástica? |
Em toda a anemia associada a manifestações do TGI ou SNC Neutrófilos plurissegmentados |
|
|
Quais são os outros achados laboratoriais da anemia megaloblástica? |
Leucopenia, trombocitopenia, elevação da LDH e bilirrubina indireta |
|
|
O ácido metilmalônico está elevado na... |
... deficiência de cobalamina |
|
|
A homocisteína está elevada na... |
... deficiência de cobalamina e folato |
|
|
A homocisteína está elevada na... |
... deficiência de cobalamina e folato |
|
|
Como devemos acompanhar a resposta à terapia de reposição com vitamina B12? |
Através da contagem de reticulócitos. Ela começa a subir por volta do 4° ou 5° dia após o início da terapia e atinge um pico no 7° dia |
|
|
Qual é a posologia de vitamina B12 a ser administrada? |
1000 mcg IM por dia por sete dias, em seguida, semanal, durante quatro semanas, e depois, mensal, indefinidamente (na maioria dos pacientes). |
|
|
Quais são as principais complicações da reposição de vitamina B12? (2) |
Hipocalemia Congestão volêmica |
|
|
Qual é a razão da diminuição do potássio? |
As células novas que estão se formando em resposta à vitamina B12 captam potássio para o seu citoplasma |
|
|
Qual é o perigo da administração de folato em pacientes com anemia megaloblástica? |
Se o diagnóstico estiver errado ou incompleto, e o paciente apresentar deficiência de cobalamina, as anormalidades hematológicas podem ser corrigidas enquanto as manifestações neurológicas não melhoram e podem progredir. Desta forma, a reposição de ácido fólico pode mascarar uma deficiência de vitamina B12 |
|
|
Se a eritropoiese medular estiver normal, com estoques de ferro, ácido fólico e B12 preservados, a meia-vida vida das hemácias pode se reduzir para até 20 a 25 dias, sem que se desenvolva anemia. V ou F? |
V - hemólise compensada Quando a meia-vida eritrocítica chega a um valor inferior a 20 dias, instala-se anemia |
|
|
Hemólise extravascular |
As hemácias são destruídas no tecido reticuloendotelial, especialmente no baço, pelos macrófagos dos cordões esplênicos de Bilroth |
|
|
Hemólise intravascular |
As hemácias são destruídas na própria circulação e seu conteúdo é liberado no plasma |
|
|
Toda hemólise crônica predispõe a formação de... |
Cálculos biliares Com maior concentração de bilirrubina na bile, podem se formar, na vesícula biliar, os cálculos de bilirrubinato de cálcio |
|
|
Diante de uma criança com episódio de crise biliar, principalmente na presença de cálculos radiopacos, até prova em contrário, o diagnóstico é de... |
Anemia hemolítica hereditária |
|
|
Reticulocitose = ? |
Aumento da produção medular de reticulócitos |
|
|
Quais anemias cursam com reticulocitose? (2) |
Anemias hemolíticas Anemia pós-hemorragia aguda |
|
|
O que são crises anêmicas? |
São as exacerbações agudas de uma anemia hemolítica crônica |
|
|
Principais condições associadas às crises anêmicas |
Anemias hemolíticas hereditárias, tais como a esferocitose hereditária e a anemia falciforme |
|
|
É a mais comum das crises anêmicas |
Crise aplásica |
|
|
Principal etiologia da crise aplásica |
Infecção pelo parvovírus B19 O vírus tem tropismo pelo pró-eritroblasto, que ao ser infectado perde a capacidade de se tornar reticulócito |
|
|
Consequência imediata da infecção pelo parvovírus B19 |
Anemia com reticulocitopenia |
|
|
Tratamento da crise aplásica |
Hemotransfusão |
|
|
O turnover aumentado de hemácias, presente nas desordens hemolíticas, exige produção aumentada de hemácias pela medula óssea e, portanto, maior utilização de nutrientes como ferro, o ácido fólico e a vitamina B12. Isso pode desencadear qual crise anêmica? |
Crise megaloblástica |
|
|
Em qual crise o paciente evolui com piora súbita da anemia, associada a um aumento da reticulocitose e da esplenomegalia? |
Crise hiper-hemolítica |
|
|
É uma crise característica da anemia falciforme em crianças < 5 anos |
Sequestro esplênico |
|
|
Sugerem anemia hemolítica (5) |
1) Leve icterícia (associada à palidez) 2) Esplenomegalia 3) História familiar positiva de anemia 4) Uso de medicamentos - hemólise autoimune (alfametildopa); hemólise não imune (dapsona) 5) Urina avermelhada ou marrom (hemoglobinúria) |
|
|
Anemias hemolíticas que mais aumentam o baço |
Talassemias |
|
|
Hemoglobinúria confirma o diagnóstico de... |
Hemólise intravascular |
|
|
Uma crise hemolítica aguda intravascular frequentemente se manifesta com... |
Febre, lombalgia, palidez, icterícia e urina escura, como no caso da deficiência de G6PD |
|
|
Enzima que está quase sempre elevada na hemólise |
Desidrogenase lática (LDH) |
|
|
Os níveis séricos de .... diminuem ou tornam-se indetectáveis na presença de hemólise |
Haptoglobina |
|
|
Indicadores de hemólise intravascular (6) |
Hemoglobinemia Hemossiderinúria Hemoglobinúria Redução dos níveis de hemopexina livre Aumento dos níveis do complexo hemopexina-heme Aumento dos níveis de metealbumina |
|
|
Tipo de anemia na hemólise |
Normo/normo Obs.: em casos de hemólise aguda grave, pode haver macrocitose (devido reticulocitose e liberação excessiva das shift cells) |
|
|
Defeitos genéticos das forças verticais (espectrina, anquirina, banda 3, proteína 4.2) levam à... |
Esferocitose hereditária |
|
|
Defeitos genéticos das forças horizontais levam à... |
Eliptocitose hereditária e suas variantes |
|
|
Fisiopatologia esferocitose hereditária |
Deficiência do ancoramento no citoesqueleto --> perda de fragmentos da camada lipidica --> esferócito (contato prolongado com macrófagos no baço) --> microesferócito --> hemólise |
|
|
Cenário diagnóstico típico da esferocitose hereditária |
Criança submetida à investigação de esplenomegalia |
|
|
Laboratório esferocitose hereditária |
Anemia micro ou normocítica HIPERcrômica Reticulócitos e microesferócitos no esfregaço de sangue periférico |
|
|
Diagnóstico esferocitose hereditária |
Anemia hemolítica crônica + reticulócitos e microesferócitos no sangue periférico + ausência de teste de Coombs positivo |
|
|
Tratamento esferocitose hereditária |
Esplenectomia!
Obs.: em crianças, ela deve ser evitada antes dos 4 anos de idade, período de maior risco de sepse fulminante por germes encapsulados |
|
|
Como ocorre a hemólise na deficiência de G6PD? |
As crises hemolíticas sempre são precipitadas por algum estresse oxidativo. O mais comum é a infecção. Diversas drogas ou substâncias com potencial oxidante também costumam desencadear as crises. |
|
|
Quadro clínico na deficiência de G6PD |
Hemólise intravascular aguda, associada a febre, lombalgia, palidez e icterícia, precipitada por uma infecção ou pela administração de uma droga (ex: dapsona, primaquina, naftaleno) |
|
|
Laboratório deficiência de G6PD |
Hemoglobinúria Hemoglobinemia Metalbuminemia Células mordidas Corpúsculos de Heinz |
|
|
Diagnóstico deficiência de G6PD |
Dosar a atividade da G6PD no sangue |
|
|
Tratamento deficiência de G6PD |
Não há tto específico. O principal é a profilaxia das crises. |
|
|
Principais condições associadas ao hiperesplenismo na prática médica (2) |
Esplenomegalias congestivas: - Cirrose hepática - Esquistossomose hepatoesplênica |
|
|
4 critérios para o diagnóstico de hiperesplenismo |
1) Citopenia de uma ou mais linhagens hematológicas (plaquetas, hemácias e leucócitos) 2) Hiperplasia reativa compensatória da medula óssea 3) Esplenomegalia 4) Correção das anormalidades após esplenectomia (este critério nem sempre é necessário) |
|
|
Anemia da insuficiência hepática grave |
Anemia hemolítica com acantócitos (excesso de colesterol na membrana plasmática) |
|
|
Mais importante anemia hemolítica adquirida |
Anemia hemolítica autoimune |
|
|
Tipos de anticorpos AHAI |
AHAI por anticorpos quentes (IgG) - contra antígenos do sistema Rh AHAI por anticorpos frios (IgM) - contra o antígeno I |
|
|
Principal local de hemólise da AHAI por IgG |
Baço |
|
|
Principais causas de AHAI por IgG (4) |
Alfametildopa LES LLC Linfomas não Hodgkin |
|
|
A associação AHAI por IgG com a PTI (Púrpura Trombocitopênica Autoimune) é conhecida como... |
Síndrome de Evans |
|
|
Confirmação do diagnóstico de AHAI |

Teste de Coombs direto |
|
|
Tratamento específico da AHAI por IgG |
Glicocorticoides (prednisona)
Se não houver resposta ou tolerância: rituximab ou esplenectomia Terceira linha: ciclofosfamida, azatioprina ou imunoglobulina intravenosa |
|
|
Principal local de hemólise da AHAI por IgM |
Fígado, por ação de seus macrófagos (células de Kuppfer) |
|
|
Quando uma causa específica de AHAI por IgM é identificada, a mais comum é... |
A infecção por Mycoplasma pneumoniae |
|
|
Forma predominante de AHAI por IgM |
Doença da Crioaglutinina |
|
|
Droga de escolha para tto da Doença da Crioaglutinina |
Rituximab 2 opção: clorambucil |
|
|
Principais drogas que causam anemia imino-hemolítica farmacoinduzida (4) |
Alfametildopa (principal) Levodopa Penicilina Quinidina |
|
|
Grandes marcos da hemólise intravascular (2) |
Hemoglobinúria Hemossidenúria |
|
|
O que são hemolisinas? |
São substâncias capazes de ativar o sistema complemento até a formação do "complexo de ataque à membrana" (C5b-C9), de modo a induzir rotura da membrana do eritrócito pela súbita entrada de água na célula |
|
|
Hemolisinas extremamente potentes (2) |
1) Anticorpos do sistema ABO 2) Veneno da aranha marrom (Loxosceles sp) |
|
|
Importante causa de hemólise intravascular grave no nosso meio |
Picada da Loxosceles sp ou aranha-marrom |
|
|
Parasitose que se comporta como uma clássica causa de hemólise intravascular |
Malária |
|
|
Doença hereditária que leva a crises de hemólise intravascular desencadeadas por estresse oxidativo |
Deficiência de G6PD |
|
|
Desordem adquirida da célula-tronco que tem como principal característica a produção de subpopulações de granulócitos, plaquetas e hemácias hipersensíveis ao sistema complemento |
Hemoglobinúria Paroxística Noturna (HPN) |
|
|
Principal causa de óbito na HPN |
Eventos veno-oclusivos
Mais clássico: síndrome de Budd-Chiari (trombose das veias supra-hepáticas) |
|
|
Diagnóstico da HPN |
Teste da citometria de fluxo |
|
|
Tratamento HPN |
Suporte |
|
|
Processo hemolítico relacionado à fragmentação de hemácias que ocorra em associação com doença em pequenos vasos, marcado por uma patologia das células endoteliais |
Anemia hemolítica microangiopática |
|
|
Principais condições relacionadas à anemia hemolítica microangiopática (8) |
1) SHU 2) PTT 3) CIVD 4) Síndrome HELLP 5) Hipertensão arterial maligna 6) Crise renal da esclerodermia 7) Carcinomatose disseminada 8) Presença de hemangiomas gigantes |
|
|
Mutação anemia falciforme |
GAG --> GTG (6-Glu --> Val) Cria o gene beta-S |
|
|
3 fatores que exacerbam o afoiçamento das hemácias |
1) Hipóxia 2) Acidose 3) Desidratação celular |
|
|
Hemoglobina mais eficiente no bloqueio à formação dos polímeros de HbS |
HbF |
|
|
2 características físico-químicas da HbS |
Tendência à polimerização Instabilidade molecular |
|
|
TODAS as células vermelhas no sangue de um falcêmico apresentam níveis aumentados de estresse oxidativo, mesmo quando não afoiçadas. V ou F? |
V |
|
|
Mecanismos de hemólise na anemia falciforme |

|
|
|
A anemia falciforme cursa com disfunção endotelial crônica, tanto na MICRO quanto na MACROcirculação. V ou F? |
V |
|
|
Resumo da cascata de eventos fisiopatológicos da anemia falciforme |
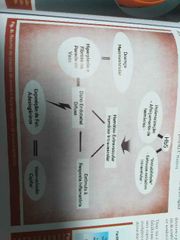
|
|
|
Laboratório anemia falciforme |
1) Queda leve a moderada da Hb e do Ht 2) Reticulocitose 3) Hiperbilirrubinemia indireta 4) Aumento da LDH 5) Queda da haptoglobina
Obs.: leucocitose e plaquetose
|
|
|
4 condutas básicas que comprovadamente diminuem a morbimortalidade da anemia falciforme |

1) Profilaxia contra o pneumococo 2) Reativação da síntese de HbF 3) Hemotransfusão 4) Uso de quelantes de ferro |
|
|
O TIBC não sofre influência direta das variações do ferro sérico. Ele representa, de forma indireta, a concentração de transferrina sérica. V ou F? |
V |
|
|
Qual é a principal causa de deficiência de B12 em nosso meio? |
Anemia perniciosa |
|
|
Qual é a principal causa de deficiência de folato? |
Alcoolismo |
|
|
Quais são as duas situações clínicas mais frequentes na hemoglobinopatia SC em relação à hemoglobinopatia SS homozigótica? |
Retinopatia e necrose de cabeça de fêmur |
|
|
Anemias hemolíticas
RESUMÃO |

|
|
|
Anemias hemolíticas RESUMÃO 2 |

|
|
|
HPN RESUMÃO |

|