Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
171 Cards in this Set
- Front
- Back
|
What limits the chemistry of a enzyme?
|
The amino acid side chains
|
|
|
What stabilizes protein structure the most? How?
|
Hydrophobic interactions; Well b/c Hydrogen bonds are necessary to get the STRUCTURE of a protein but hydrophobic interactions stabilize the structure
|
|
|
What characterizes protein structure?
|
1. formation of regular secondary structure
2. H-bonds involving backbone and side-chain atoms 3. burial of non-polar side chains to form hydrophobic core 4. Stable structure that is in motion around a central core (like the crust of the earth moving around the Fe core) |
|
|
What is Aspartame made of?
|
Aspartate and phenylalanine and a methyl ester
|
|
|
When does tyrosine become an essential amino acid?
|
When it is not able to be transported into the brain; normally phenylalanine and tyrosine are transported into the brain via the same transporter, but if you have PAH deficiency suddenly tyrosine will become an essential A.A.
|
|
|
What does tetramerization allow?
|
Tetramerization domain allows for dimers and trimers to form => quaternary structure
|
|
|
What happens in classic PKU?
|
The active site will not be available at all, H285 binds required iron atom in the active site but a mutation to H285 will cause the enzyme to not be able to bind Fe and so Classic (full-fledged) PKU will result
|
|
|
What is required as a cofactor for PAH?
|
BH4
|
|
|
What dz will result from BH4 deficiency?
|
The dz will present like PKU but it is easily solved if you give the pt BH4 because then they will be right back to normal
|
|
|
What does a mutation of valene to methionine cause?
|
Variant PKU b/c of the dec. affinity for phenylalanine and BH4, V388M is important for active site functionality
|
|
|
What is phenylalanine w/ respect to PAH?
|
Phenylalanine is an allosteric effect and substrate, phenylalanine is a competitive inhibitor for tyrosine on PAH
|
|
|
What effect will a mutation to the regulatory site of PAH have?
|
It will prevent activation or shift activity curve
|
|
|
What effect will a mutation to an enzymes phosphorylation site have?
|
It will remove the regulatory mechanism
|
|
|
What are most important for enzyme function?
|
Functional residues (A.A.)
|
|
|
What causes Marble Brain Syndrome?
|
A mutation in CA II; causes the brain to become calcified (turned to marble)
|
|
|
What influences the activity of an enzyme?
|
Temperature
|
|
|
What specific mutation causes cystic fibrosis?
|
A mutation to a single phenylalanine residue
|
|
|
What is the most common class of cystic fibrosis?
|
Class II: a defective maturation and premature degradation of the protein
|
|
|
What are kinetic traps?
|
Due to cell crowding, proteins can fold into local energitic minima that are not the right structure => a kinetic trap
|
|
|
How do chaperones work?
|
They recognize kinetic traps and exposed hydrophobic residues -> unfold proteins -> refold correctly or prevent trap from occurring
|
|
|
What will happen if the chaperone cannot bind to a protein?
|
It will bind to a proteasome and then degrade the protein
|
|
|
What do ribosome-associated chaperones do?
|
Trigger factors protect small proteins or single domains => creates a protective environment to help the protein fold
|
|
|
What do hydrolases do?
|
Use water to cleave a bond
|
|
|
What do lyases do?
|
Just cleave a bond w/o using H2O, this is what bacteria use
|
|
|
What is a mucopolysaccharidosis?
|
A deficiency of lysosomal enzymes that degrade GAGs
|
|
|
Where does cathepsin come from?
|
Lysosome
|
|
|
Where are lysosomal proteins synthesized? Where do they go then? and after that?
|
Proteins in the lysosome are synthesized in the ER, then go to the Golgi then to the LATE endosome
|
|
|
What is the difference b/e early endosomes and lysosomes?
|
Early endosomes come from the membrane and lysosomes have a drop in pH and presence of unique enzymes
|
|
|
How are hydrolases targeted to the lysosome?
|
N-linked => attached to asparagine residue => and mannose residue is phosphorylated which then attaches to a mannose-6-phosphate receptor
After binding to M6P receptor, clathrin will bind and coat the vesicle which will then break off and fuse w/ lysosome |
|
|
What is type II pompe dz?
|
The only glycogen storage dz that is a lysosomal storage dz
|
|
|
How are GAGs degraded?
|
Must first be engulfed then fuse w/ a lysosome:
>oncloglycosidis produce oligosaccharides where degradation happens from the end so the parts added last will get degraded first |
|
|
What causes mucopolysaccharidosis? what does this dz lead to?
|
A lysosomal hydrolase deficiency that is involved in heparin or dermatan sulfate degradation
Leads to inc. glycosaminoglycans -> which are found in pee |
|
|
What dz'es are mucopolysaccharidosis dz'es?
|
1. Hurler
2. Hunter 3. SLY 4. San Filippo |
|
|
What do phospholipids need in order to be degraded?
|
phospholipase or sphingomyelinase
|
|
|
What causes Neimann-Pick dz?
|
Degradation of sphingomyelin deficiency
|
|
|
What do lysosomal enzymes degrade primarily?
|
Degrade primary extracellular proteins and cell-surface membrane proteins that are used in receptor-mediated endocytosis
|
|
|
What is I cell dz? Where will stuff accumulate?
|
When you are deficient in the enzymes that are needed for M6P formation=> if you cannot traffic the hydrolase properly by M6P they will be secreted into cell cytosol
|
|
|
What steps are required for formation of M6P?
|
UDP-acetylglucosamine: lysosomal enzyme N-acetylglucosamine-1-phosphotransferase
N-acetylglucosamine 1-phosphodiester-N-acetylglucosaminidase |
|
|
What does a deficiency in enzyme N-acetylglucosamine-1-phosphotransferase or N-acetylglucosamine 1-phosphodiester-N-acetylglucosaminidase result in?
|
An I-cell dz: b/c hydrolase will not be trafficted to lysosome, so hydrolases will float around in the cytosol w/ no target, but this is not fatal b/c they are not at their optimal pH for full functioning
|
|
|
What do extracellular hyaluronidases do? (Hyal-2)
|
Make smaller chains by hydrolysis
|
|
|
What do HARE receptors do?
|
They are hyaluronan receptors for endocytosis that recognize smaller chains and cause endocytosis
Endosomes w/ hyaluronan fuse w/ the lysosome |
|
|
What do hyaluronidases do? where is it located?
|
1. Hydrolize hyaluronan into pieces by cleaving beta (1,4) bonds
2. excreted enzyme so it is extracellular and is linked to a lipid -> will be on the plasma membrane |
|
|
Where are Hyal-2 and Hyal-1 found?
|
Hyal-2: found extracellular and GPI linked
Hyal-1: in the lysosome, also in serum |
|
|
What is hyaluronan catabolism like?
|
Occurs in lysosome, the terminal sugar is removed from the non-reducing end of hyaluronan fragments by exoglycosidases
Hyaluronan fragments inside lysosome are broken down beta-glucoronidase and beta-N-acetylglycosaminidase who finish degradation to monosaccharides which leave the lysosome and are re-usable |
|
|
What does beta-glucoronidase do?
|
Cleaves glucuronic acid
|
|
|
What does beta-N-acetylglucosaminidase do?
|
Cleaves N-acetylglucosamine
|
|
|
What enzyme breaks down the repeating disaccharides in hyaluronan?
|
Beta-glucuronidase and beta-N-acetylglucosaminidase
|
|
|
Where is antithrombin synthesized?
|
In the liver
|
|
|
What does antithrombin do?
|
It is a serine protease inhibitor that binds and inactivates thrombin, Factor Xa and Factor IXa which are required for homeostasis (coagulation)
|
|
|
What does the anti-coagulant heparin do?
|
Binds to antithrombin and accelerates the rate of antithrombin inactivation of serine proteases
*basically makes anti-coagulation happen faster |
|
|
What is the function of heparanase?
|
>Degrades the basement membrane
>Procoagulant activity So cancer cells that overexpress Heparanase are more metastatic |
|
|
What happens during heparan sulfate catabolism?
|
Acetyl transferase in lysosomal membrane needs Acetyl CoA to do the reaction, then the sulfate is removed first
|
|
|
What does chondroitan sulfate have in it?
|
Glucoronate
|
|
|
What does dermatin sulfate have in it?
|
A iduronic acid
|
|
|
Where does glucosamine tend to be found?
|
IN chondroitan instead of keratin sulfate
|
|
|
Where does mitophagy happen? What is it?
|
Occurs in pre-RBC cells, it is the fusion of the mitochondria w/ the lysosome
|
|
|
What is the charge of the erythrocyte glycocalyx?
|
Negative or neutral
|
|
|
what limits the attraction btw RBCs and endothelial cells?
|
Carbohydrates on the RBC membrane as well as membrane proteins
|
|
|
When are spectrin and ankyrin added to the membrane?
|
They are minimally present on the membrane until Band-3 and protein 4.1 are synthesized
|
|
|
What mutation will be caused by a mutation to spectrin alpha or beta?
|
Spherocytosis
|
|
|
What will happen in decreased spectrin synthesis or dec. ankyrin synthesis?
|
Leads to poor binding of molecules which lack binding will decrease spectrin recruitment and weaken verticle interactions => this leads to microvesicles being popped off=> dec. RBC surface area => leads to dec. O2 and CO2 exchange
|
|
|
What are ABO Ags?
|
Mostly carbohydrates that are part of RBC membrane, they are basically a part of the glycoprotein coat
They are the most reactive Ag on the RBC membrane |
|
|
What is the difference b/e Type A, B and O blood types?
|
A has an N-aceteal linkage
B does NOT have a N-aceteal linkage and O has an enzyme but it does not work |
|
|
What are stealth RBCs?
|
They are Ags that are modified to hide via carbohydrates, they shield their proteins w/ sugar so Abs cannot bind and attack the blood
|
|
|
What does 2,3DPG preferentially bind to?
|
deoxyHb or RBCs in the T state
|
|
|
What is synthesized by glycolysis per glucose molecule?
|
4 ATP (2 net ATP), 2 NADH and 2 pyruvate from 1 glucose and 2 NAD+ and 2 ATP
|
|
|
What would accumulate in triosephosphate isomerase deficiency?
|
dihydroxyacetone phosphate
|
|
|
What effect does oxidant stress have on RBCs?
|
High levels of oxidant stress lyses RBCs due to membrane damage (causing ion leakage) and proteins (loss of enzyme activity)
|
|
|
What effect does oxidizing a thiol residue in GSSG have?
|
Will deactivate the enzyme b/c thiol will participate in the chemical event
|
|
|
What does glutathione peroxidase use in its reaction?
|
Will take glutathione and oxidize H2O2 to water and GSSG using NADPH
|
|
|
What deficiency is protective against malaria?
|
G6P dehydrogenase deficiency b/c cells can't handle the stress of malaria invasion and lyse and trigger the immune system before the bacteria can invade
|
|
|
What does xanthine oxidase do?
|
makes superoxide which reacts w/ NO to form ONOO-; which will destroy anything around it
|
|
|
How can vitamin C act as a pro-oxidant?
|
By converting Fe(III) to Fe(II) which is good BUT then the Fe(II) can be reconverted to Fe(III) and produce another OH*
|
|
|
What reduces methemoglobin?
|
Cytochrome b5 (NADH) and methemoglobin reductase reduce methemoglobin
|
|
|
What will band 3 protein (CDB3) bind to in high oxygen situations? low oxygen situations?
|
High O2: CDB3 will bind to PFK and force glucose to go to the PPP and make NADPH for anti-oxidant purposes
Low O2: CDB3 will bind to deoxy Hb and release PFK, so glucose will now undergo glycolysis |
|
|
How do neutrophils, monocytes/macrophages kill bacteria? How do Eosinophils kill bacteria?
|
Using HOCl and OH*, they have myeloperoxidase
Eosinophils generate HOBr |
|
|
What does myeloperoxidase do?
|
Makes bleach HOCl which
|
|
|
How are cholesterol deposits broken down?
|
Removed by macrophages and packed w/ apolipoprotein A1, activation of macrophages produces ROS (HOCl)
HOCl damages apoprotein A1 preventing removal of cholesterol => so macrophages actually CANNOT remove cholesterol |
|
|
What is ataxia?
|
A pyruvate dehydrogenase deficiency, so not enough ATP will be made
|
|
|
What are the three enzymes of the pyruvate dehydrogenase complex?
|
1. pyruvate decarboxylase
2. dihydrolipoyl transacetylase 3. dihydrolipoyl dehydrogenase |
|
|
What is the E1 (pyruvate decarboxylase) enzyme? what does it do? what is a biproduct of its reaction? what cofactor does it require?
|
Composed of a tetramer w/ 2 alpha and 2 beta that requires thiamine (B1) as a cofactor that converts TPP to thiamine pyrophosphate which release CO2 as a biproduct
|
|
|
What is E2 (dihydrolipoyl transacetylase)? what does it do? what cofactor does it require?
|
A monomer w/ a long flexible chain that picks up actetylTPP and transfers it to CoA to make acetyl CoA; links to lipoic acid which is joined to a lysyl residue through amide links
It oxidizies the active acetaldehyde to make acyl group which is transferred to CoA |
|
|
How does arsenic affect the PDH complex?
|
Arsenic binds to reduced lipoic acid and makes it unavailable to work, so administer a chelating agent to get rid of it
|
|
|
What is E3? (dihydrolipoyl dehydrogenase) What does it do? what cofactor does it require?
|
A monomer that oxidizes lipoic acid by reducing FAD to FADH2, it then reduced NAD+ to NADH by using FADH2. It needs Riboflavin (B2) to reduce FADH2 to FAD+ and Niacin (B3) to reduce NAD+ to NADH
|
|
|
What effect does excess intake of riboflavin (B2) have? deficiency?
|
Excess: causes yellow urine and interferes w/ B1 and B6
Deficiency: leads to neurologic problems |
|
|
What does Niacin (B3) do in the PDH complex? What does a deficiency of Niacin cause?
|
Niacin is needed to convert NAD+ to NADH; a deficiency causes pellagra, but tryptophan can serve as an alternative source
|
|
|
What is the function of panthothenic acid (B5)
|
Important for acetyl CoA; w/o pantothenic acid (B5) Acetyl CoA cannot be made
|
|
|
What does PDH kinase do? what triggers it?
|
Converts PDH to inactive form; which is triggered by inc. ATP, inc. NADH, dec. pH, inc. acetyl CoA and inc pyruvate
|
|
|
What does PDH phosphatase do? what triggers it?
|
Converts PDH complex to active form via Ca
|
|
|
What regulates PDH kinase/phosphatase?
|
phosphorylation; phosphorylation will inhibit the enzyme
|
|
|
What does biotin do? where is it synthesized? what does it do?
|
Active in bond making and forming in enzyme especially in TPP and B5, it is synthesized by intestinal bacteria inside you
|
|
|
How does large amounts of EtOH effect the cell?
|
Affects NADH/NAD+ ratio, by making a lot of NADH it turns PDH A (active) into PDH B (inactive) which backs the system up and results in lots of storage of sugars and a fatty liver
|
|
|
What is the glycerophosphate shuttle?
|
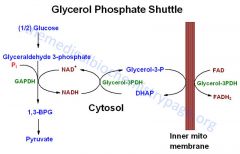
Source of H+, through FADH2 => which feeds directly into the ETC
|
|
|
What does the malate-aspartate shuttle do?
|
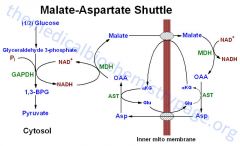
Shuttles NADH to the matrix
|
|
|
How does the malate-aspartate shuttle work?
|
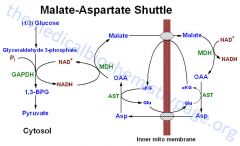
OAA to malate (which cross inner mitochondrial membrane) to OAA => generates one NADH, the H+ is carried across by malate/a-KG BUT you need to transfer OAA back across the membrane or the cycle will stop
|
|
|
How does the malate-aspartate shuttle retransport OAA across the innermitochondrial membrane?
|
By converting aspartate back to OAA
|
|
|
What does malate dehydrogenase work?
|
In the cytosol: converts NADH -> NAD+
In inner mito membrane: converts NAD+ -> NADH |
|
|
Where does the glycerophosphate shuttle feed into the ETC?
|
In complex II where FADH2 is used
|
|
|
What is the final electron acceptor in the ETC?
|
O2
|
|
|
What is coenzyme Q?
|
Similar to ascorbic acid so it has antioxidant capabilities, it can transport one or two electrons at a time; it is a hydrophobic molecule that floats in the membrane
|
|
|
What complexes in the ETC are most likely to generate ROS?
|
complexes 1 and 3 b/c they are the most leaky
|
|
|
Why are mitochondria the most likely organelle to be mutated?
|
B/c they do not have the PPP (which is the in the cytosol); the mito balances this by having multiple copies of genome and lots of mitos in the cell
|
|
|
What inhibits ATP synthase?
|
Oligomycin
|
|
|
How can ATP synthase make a proton gradient?
|
By hydrolyzing ATP if there is loss of H+ gradient; this occurs in hypoxia or cyanide poisoning
|
|
|
What effect does salicylate toxicity have? (Aspirin)
|
Salicylate can cross membranes even though it is charged b/c it can make H-bonds b/e adjacent groups; but it is still acidic so at high concentrations it will disrupt the ability to make ATP by destroying the H+ gradient
|
|
|
What enzymes in the TCA release CO2?
|
Isocitrate dehydrogenase and alpha-ketoglutarate dehydrogenase
|
|
|
What is the rate-limiting step in the TCA?
|
citrate synthase
|
|
|
What enzymes have a neg. delta G?
|
citrate synthase, isocitrate dehydrogenase and a-KG dehydrogenase
|
|
|
What inhibits the enzymes of the TCA?
|
high [NADH] and high [ATP]
|
|
|
What does lipoprotein lipase do?
|
Hydrolyzes chylomicrons to free fatty acids and glycerol
|
|
|
What do lipolytic hormones do?
|
Signal tissue to release more fatty acid
|
|
|
What signaling mechanism do lipolytic hormones use?
|
AC => cAMP => PKA cascade
|
|
|
What activates hormone sensitive lipase?
|
PKA phosphorylation
|
|
|
What are fatty acids bound to in blood?
|
Albumin
|
|
|
What does PKA do when activated by lipolytic hormones?
|
phosphorylates porillipin protein whcih provides a docking place for adipose tissue triglyceride lipases which take fat and start to hydrolyze the fat
|
|
|
What are the steps to hydrolyze stored fat starting from the hormone binding?
|
Lipolytic hormones bind to G-protein receptor => AC->CAMP->PKA => PKA activates hormone sensitive lipase and porillipin => porillipin provides a docking place for adipose tissue triglyceride lipase to start hydrolyzing fat => hormone lipase hydrolyzes fat even more => then monoglyceride hydrolase will finish the job and release to surrounding tissue
|
|
|
What are the products of stored fat hydrolation?
|
A glycerol (which is a good gluconeogenic substrate) and fatty acids
|
|
|
What are the products of beta-oxidation? where does it occur?
|
products are acetyl CoA, FADH2 and NADH (acetyl CoA is oxidized to CO2 and H2O in the TCA cycle
Happens in the mitochondria |
|
|
Where are fatty acids activated? by what?
|
Fatty acids must be activated before they are metabolized which occurs in the cytosol by fatty acyl-CoA synthetase (which uses ATP)
|
|
|
What does carnitine-acylcarnitine translocase do?
|
Transfers FA into the matrix and also transfers carnitine to the inner mito space
|
|
|
What does each turn of beta-oxidation yield?
|
14 ATP and each turn makes the fatty acid 2 carbons shorter
|
|
|
What is the basic structure of the beta-oxidation pathway?
|
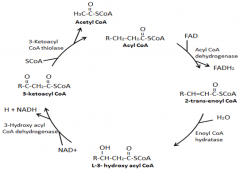
|
|
|
What inhibits carnitine acyltransferase I?
|
Malonyl CoA by blocking the substrate getting into mito.
|
|
|
What inhibits beta-hydroxyacyl CoA Dehydrogenase?
|
![High [NADH]](https://images.cram.com/images/upload-flashcards/90/77/48/2907748_m.png)
High [NADH]
|
|
|
What inhibits thiolase?
|
![High [Acetyl CoA]](https://images.cram.com/images/upload-flashcards/90/77/45/2907745_m.png)
High [Acetyl CoA]
|
|
|
What cofactors are required for oxidative phosphorylation to occur?
|
ADP and Pi
|
|
|
What effect would diminishing the substrate for the anaplerotic reactions have?
|
It would slow the TCA cycle => Inc. [Acetyl CoA] => ketogenesis
|
|
|
What is the function of MPF? (Cyclin b-cdc2/cdc)
|
Reversible breakdown of the nuclear envelope and role of nuclear lamin proteins via DNA transfaction experiments
|
|
|
What do SMS (structural maintenance proteins) do?
|
Required for normal chromosome segregation, they are called condensins
|
|
|
What effect does phosphorylation of condensins have?
|
Necessary for maintaining the condensed state of the chromosomes via DNA binding
|
|
|
What does phosphorylation of APC result in?
|
initiation of anaphase
|
|
|
How is anaphase induced?
|
By degradation of the anaphase inhibitor (securin) via the APC pathway activated by MPF
|
|
|
What does securin do?
|
Stabilizes the cohesins to prevent them from separating; cohesins act as linchpins to tie sister chromatids together
|
|
|
What is the effect of degradation of mitotic cyclin B? when does this happen?
|
Degradation occurs via APC pathway late in anaphase, the cyclin must be degraded in order for anaphase to take place
|
|
|
What does M cyclin-CDK do?
|
Initially responsible to keep securin-separase complex stable and inactive so it needs to be degraded by APC for separase to cleave cohesin
|
|
|
When does regulated degradation of mitotic cyclins occur? how does it occur?
|
Occurs late in anaphase via the APC-ubiquitin pathway which makes use of a destruction box common to the N-T of mitotic cyclins
|
|
|
What does cyclin activation require?
|
2 STEPS: cyclin binding and then phosphorylation by CAK
|
|
|
Whose activity is reduced in G0 (quiescent) cells? whose activity is elevated in tumor cells?
|
CAK activity is decreased in quiescent cells and elevated in tumor cells
|
|
|
What is the sequene of cyclin-cdk activation in mitosis?
|
cyclin B => CDK1 => APC
|
|
|
What do INK4 proteins do?
|
Bind to CDK4/6 to block cyclin D binding
|
|
|
What do Cip/Kip do?
|
block CDK2-cyclin E but they also bind to and block active site of multiple CDKs
|
|
|
What do CKIs do?
|
Regulate entry into S phase => G1-S transition
|
|
|
What is the action of mitogens?
|
They activate the Cyclin D-CDK 4/6 complexes, inhibit CKIs
|
|
|
What does Rb inhibit?
|
E2F
|
|
|
What does E2F do?
|
a transcription factor of Cyclin A and D
|
|
|
What will happen if Rb is phosphorylated (inhibited)?
|
It will not block E2F, so E2F will keep influencing the cell cycle to continue
|
|
|
When is Rb normally activated?
|
Not until mid/late G1 phase when the buildup of CDK2-cyclin E and CDK 4/6 -Cyclin D leads to phosphorylation of Rb and release of E2F from suppression
|
|
|
What arrises due to DNA damage?
|
p53 protein which stabilizes DNA damage => chk2 will phosphorylate it and make it less susceptible to degradation
|
|
|
What happens when p53 is activated?
|
It will activate transcription of the CDK inhibitor CKJ which binds to all CDKs and inhibits their actions, which prevents the cell from moving through the cycle until the DNA damage has been repaired
|
|
|
What does p53 due if the DNA damage is too severe to be fixed?
|
p53 activates genes that lead to apoptosis via BCL-2
|
|
|
What is folate used for?
|
Used to make THF
|
|
|
What does thymidylate synthase do?
|
Catalyzes the conversion of dUMP to dTMP
|
|
|
What does catabolism of Purines create?
|
Uric acid which will make crystals and cause Gout
|
|
|
What effect does excess PRPP have?
|
Leads to increased purine synthesis and subsequent degradation of the extra purines which results in inc. uric acid and thus gout
|
|
|
What does a deficiency of HGPRT cause?
|
Lesch-Nyhan syndrome (which is characterized by pt desires to self-mutilate)
|
|
|
What does Adenosine deaminase deficiency cause (ADA)?
|
Severe combined immunodeficiency dz (SCID) which causes a build-up of dATP which inhibits ribonucleotide reductase and prevents the synthesis of the other dNTPs
|
|
|
What does Glycosylase do?
|
Flips base into active site to recognize a mismatch => glycosylase has some specificity b/c when you have incorrect base pairing you tend to have weak connections that could cause the damaged bp to be easily taken up by glycosylase
|
|
|
What does DNA polymerase beta do?
|
used for repair => not same as replication pool
|
|
|
What does AP endonuclease do?
|
Removes sugar and phosphate
|
|
|
What does oxidation of guanine cause?
|
Leads to purine-purine base pairing, 8-hydroxyguanine acts like thymine so bps w/ adenosine
|
|
|
What does Mut Y do?
|
Recognizes purine-purine base pairing and takes away "A" from daughter cells
|
|
|
What does Mut T do?
|
Takes away 8-hydroxyguanine => makes T-A bp
|
|
|
What inhibits thymidylate synthase?
|
fDUMP
|
|
|
What effect does inc. dUTP concentration and dec. dTTP concentration have?
|
results in putting uricil in DNA, polymerase will take uricil out of DNA so now you have an a-basic site
|
|
|
What is the effect of O6-methylguanine cause?
|
Causes methylation of guanine and base pairing w/ thymine instead of cytosine which results in G-C -> A-T transitions
|
|
|
What is the only DNA repair enzyme capable of direct repair?
|
O6-MGMT b/c it will go and remove the methyl group from O6-Mg
|
|
|
What effect does UV damage have on DNA?
|
Will cause crosslinks btw itself and will cause DNA damage => pyrmidine-pyrimidine linkage
|
|
|
How are thymine cross-links repaired?
|
By nucleotide excision repair
|
|
|
What effect does ionization damage have on DNA?
|
Causes a double-strand break that occurs when DNA damage or gaps are on both strands
|
|
|
How are double-strand breaks in DNA fixed?
|
By homologous recombination
|
|
|
Why is mismatch repair more error prone that nucleotide excision repair?
|
B/c unlike excision repair you do not need a specific glycosylase to recognize which of the two bases is wrong
|