Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
76 Cards in this Set
- Front
- Back
|
______________: Multiple deep, yellow lesions on Bruch’s membrane & RPE. Causes congenital stationary night blindness, with otherwise normal visual function. Usually born with, and does not get better as pt age.
Suspected autosomal recessive inheritance. |
Fleck Retina Syndromes
|
|
|
ERG findings of Fleck Retina of Kandori
EOG findings? |
Delayed, but eventually normal (after about 3 hours) dark adaptation thresholds and scotopic ERG amplitudes once the patient is dark-adapted for sufficient amount of time
EOG NORMAL!! |
|
|
Fleck Retina of Kandori lesion
|
White, cloud-like lesions at the level of the pigmented epithelium extending from macula out to equator.
|
|
|
What is the prognostic significance of a diagnosis of fundus albipunctatus vs. retinitis punctata albescens?
|
Answer: Fundus albipunctatus is a form of congenital stationary night blindness and would not be expected to produce progressive visual loss. Retinitis punctata albescens acts clinically like retinitis pigmentosa and results in progressive visual field loss, night-blindness, and retinal vascular attenuation.
|
|
|
______________:Discrete uniform white dot-like lesions, most in mid-periphery of both eyes.
Autosomal recessive.. Onset first to second decade (early onset). Congential, stationary condition that does not get better throughout life. Complaints of nyctalopia (night blindness). |
. Fundus Albipunctatus (subtype)
Form of congenital stationary night blindness in which patients are night-blind for life. |
|
|
Fundus Albipunctatus (subtype)
Fundus findings: |
a. Discrete uniform white dot-like lesions, most in mid-periphery of both eyes.
b. Can be stationary, can increase, or can regress. c. Normal caliber retinal vessels, and macula appears normal 1) DDx: Compare to RP, which has narrowed vessels. |
|
|
Fundus Albipunctatus EOG and ERG findings
|
vi. ERG:
a. Normal in some. b. Reduced scotopic ERG response is typical. If you let the patient dark-adapt for several hours, the scotopic ERG will be normal. c. Regeneration of scotopic ERG waveform with prolonged dark adaptation in 1 to 12 hours. vii. EOG: normal or abnormal (parallels ERG findings). viii. Visual fields: normal. |
|
|
____________:
Exactly like Retinitis pigmentosa with white flecks. ii. Unlike fundus albipunctatus, this condition is progressive. This is not a stationary disease. |
2. Retinitis Punctata Albescens (subtype)
|
|
|
T/F Fundus albipunctatus is a PROGRESSIVE disease
|
FALSE, stationary
|
|
|
T/F Reinitis Punctata Albescens is a PROGRESSIVE dsease
|
T
|
|
|
_________________: Same disease due to the fact that you tend to get both conditions in the same eye)
One of the most commonly seen inherited macular dystrophies. |
Fundus Flavimaculatus and Stargardt’s disease (Fundus flavimaculatus: white “flecked retina”; Stargardt’s: “macular lesion”
|
|
|
Fundus Flavimaculatus and Stargardt’s disease have what type of hereditary pattern?
|
autosomal recessive (90%)
|
|
|
Onset of Fundus Flavimaculatus and Stargardt’s disease
|
Onset somewhere between ages 6-20, although visual impairment may not be apparent until as late as ages 30 to 40. VA decrease will precede any changes noted at the macula!
|
|
|
VA typical of Fundus Flavimaculatus and Stargardt’s disease
|
Asymptomatic if no macular lesion.
Decreased central vision in patient with previously normal vision NOTE: Visual loss frequently precedes macular findings!!!! iii. Moderate color vision defect. |
|
|
Fundus Flavimaculatus and Stargardt’s disease Fundus appearance
|
ii. Unique, peculiarly shaped, angulated, linear yellow-white flecks(pisciform, “fish-tail”) within posterior pole and mid-periphery (called fundus flavimaculatus when present without a macular lesion)
a. With time the yellow color fades, and is replaced by pigmentation surrounded by whitish halos, or “salt and pepper” pigmentary changes (stippling). iii. “Beaten metal” ovoid patch in macula (Stargardt's disease)- looks like a black eye: |
|
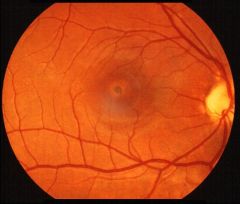
What disease is this?
|
“Beaten metal” ovoid patch in macula (Stargardt's disease)- looks like a black eye:
|
|
|
Presence of mid-peripheral flecks, especially early in life, carries a poorer visual prognosis for patients with __________ (vs. when the disease is limited to the macula).
Diagnosis is made by bilateral symmetric macular lesions centrally with surrounding flecks. DDx: If this is seen in a 70 year old, consider age related macular degeneration. But for this picture in a young patient, think of ____________________- |
Stargardt’s disease
fundus flavimaculatus and Stargardt’s disease. |
|
|
FA of ______________________:iv. May have "dark (silent) choroid" (as a result of excessive lipofuscin filling up RPE cells to give it a plush orange carpet appearance).
No choroidal fundus glow, because the RPE is infiltrated by lipofuscin that blocks the choroid. |
Stargardt's or fundus flavimaculatus
|
|
|
_________________________: Perhaps one of the most common hereditary disorders.Autosomal dominant with variable penetrance.Onset second to fourth decade
|
Familial drusen (Hutchinson-Tay choroiditis, Holthouse-Batten superficial chorioretinitis, Doyne's honeycomb dystrophy, malattia leventinese, Dominant Hereditary Drusen)
. |
|
|
___________ pts: These patients are fine for most of their lives, unless they develop a disciform lesion
Disciform lesion may develop later in life due to choroidal neovascularization, or earlier due to fluid that develops under macula. 4. Asymptomatic unless disciform detachment (disciform: fluid present under the macula causing elevation; usually not associated with CNV |
Familial Drusen
|
|
|
In Familial drusen, a ______________ may develop later in life due to choroidal neovascularization, or earlier due to fluid that develops under macula.
|
Disciform lesion
|
|
|
FA of Familial drusen
|
6. Fluorescein angiography:
i. Transmit or block early, with eventual stain (“starry sky” angiogram). DDx: Drusen do not leak. |
|
|
Familal drusen symptoms
|
In third or fourth decade patients may notice:
* decreased visual acuity * metamorphopsia * paracentral scotoma |
|
|
What color defect is associated with familial drusen?
|
Color vision testing: mild tritan defect.
|
|
|
Benign Familial Fleck Retina is an autosomal __________ disorder characterized by ______________. Pt is usually asymptomatic and will have _______ visual function. Fluorescein angiography reveals irregular __________________ suggesting a diffuse abnormality of the _________
|
A. Autosomal recessive disorder B.diffuse white-yellow (larger and more uniform) flecks throughout the fundus with relative sparing of the macula.
C.Asymptomatic, and have normal visual function. D. hyperfluorescence that does not correspond with the distribution of flecks, suggesting a diffuse abnormality of the RPE. |
|
|
________________: Onset in first decade in most:
Occasionally not diagnosed until much later. Fundus findings precede visual loss Very dramatic fundus changes! |
Best Disease/Vitelliform Dystrophy
|
|
|
DDx Stargardts vs, Best disease
|
In Best Disease, Onset in first decade in most, though might not be diagnosed until much later.
becauseFundus findings PRECEDES visual loss a. DDx: Opposite of Stargardts which has VA loss before you ever see anything in fundus! |
|
|
Is Best's Disease
1, Bilateral? 2. Symetrical? |
Fundus findings
Usually bilateral, symmetrical, and central |
|
|
Previtelliform stage of Best's Disease
|
(no egg yolk seen):
i. Stage 0: Normal fundus with abnormal EOGs. ii. Stage I: Pigment mottling in macula. Cannot tell what disease can cause mottling without further testing at this point |
|
|
Stage 2 of Best's Disease:
|
Vitelliform stage (classical stage):
Egg-yolk lesion in fovea a. Single or multiple lesions. Lesions are not always yellow in color. b. Bilateral and symmetrical. c. Consistent with 20/20 vision, even with macula appearing to be poor. VA may fluctutate. d. FA shows marked hypo-flourescence in zone covered by lesion early. hyperfluorescence later in this stage. |
|
|
____________ stage of Best's Disease involves Orange-tinged discoloration to egg-yolk appearance as it breaks down.
b: Partial resorption of cystic fluid . "Pseudohypopyon stage". c: Total resorption of cystic fluid without atrophy of the choriocapillaris. Followed by atrophy of the choriocapillaris; bilateral and symmetrical. |
Stage III: Vitelleruptive Stage; egg yolk breaks up
|
|
|
Stage 4 of Bests Disease
|
7. Stage IV: Scarring stage:
i. Stage IVa: Resorption of the majority of cystic fluid and presence of a fibrotic-appearing scar (“Scrambled egg"). ii. Stage IVb: Stage IVa with choroidal neovascularization and fibrovascular scar or appearance of subretinal hemorrhage. |
|
|
T/f VA DECREASES as Stages of Best's Disease increases
|
T
|
|
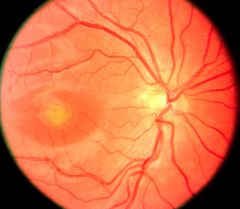
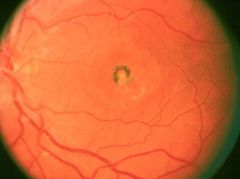
What disease is this:
What stage is this? |
Classic egg-yolk appearance in the second (vitelliform) stage of vitelliform macular dystrophy
|
|
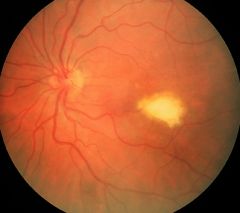
Stage 3 of Best's disease involves what?
|
The scrambled egg appearance results from a deterioration of the uniform cystic lesion noted in stage 2 (egg-yolk appearance). At this point, the visual acuity can begin to worsen.
The pseudohypopyon (stage 3) lesion is found in the teenage or later years. It results from a break in the retinal pigment epithelium, allowing accumulation of the yellow substance in the subretinal space with the formation of a fluid level. |
|
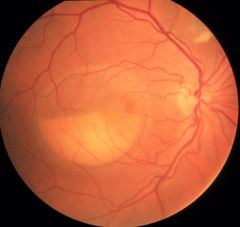
What disease is this?
|
Best's Disease:
The atrophic stage, scarring stage (stage 4) may be accompanied by the deposition of pigment or choroidal neovascularization, both of which can lead to visual deterioration. |
|
|
What is the ONE test that tells you your pt has Best's?
|
****Electrophysiology****
1. ERG normal. 2. EOG abnormal in affected patients and in carriers. This is one of the most useful tests in confirming the diagnosis, because usually if the ERG is normal, the EOG is also normal. Therefore, one test done to see if a patient has Best’s disease is EOG. Will probably be a test question. |
|
|
_______________:Onset fourth to sixth decade (in older patients).Normal vision to slight visual decrease ( 0/30-20/60)
May have metamorphopsia. 1. Symmetrical, round or oval, slightly elevated, yellow sub-retinal lesion 1/3 disc diameter in size (up to 1 DD). 2. Variable central pigmentation (dark spot in the center). |
Adult Onset Foveomacular Vitelliform Dystrophy (aka Peculiar foveomacular dystrophy of Gass, Adult-onset foveomacular pigment epithelial dystrophy (AOFPED), Pseudovitelliform macular degeneration, Adult vitelliform macular degeneration, Pseudo-Best’s disease (often confused with Best’s))
|
|
|
Only Pattern dystrophy that is NOT autosomal dominant?
|
Exception: Sjogren's reticular is autosomal recessive.
|
|
|
Pattern Dystrophy fundus finding
|
. Yellow or black pigmented pattern at level of RPE (multifocal, similar in appearance to Flavimaculatus).
Name describes changes at level of RPE HCI. Bilateral, symmetric |
|
|
______________dystrophy: Point mutation in codon 167 of the peripherin or RDS (retinal degeneration - slow) gene on the chromosome. If you had a different defect in the SAME gene, you would have Retinitis Pigmentosa or Retinitis Ablicans
|
Butterfly-pattern
|
|
|
Other Terms for Retinitis pigmentosa include:
|
Retinitis pigmentosa (and allied disorders)
A. Synonymous terms used in the literature over the years (Dr. Deglin said to know these!): 1. Tapeto-retinal pigmentosa. 2. Primary pigmentary retinal degeneration. 3. Pigmentary retinopathy. 4. Rod-cone dystrophy |
|
|
__________: characterized by inherited, progressive dysfunction, cell loss, and eventual atrophy of retinal tissue
i. Initial involvement of photoreceptors. ii. Subsequent damage to inner retinal cells. |
Retinitis Pigmentosa
|
|
|
RP + deafness is known as
|
Usher's
|
|
|
________________: HALLMARK symptom of RP
|
Nyctalopia (night blindness) –
|
|
|
Congenital stationary night blindness differentials:
|
• Oguchi’s disease:_diffuse yellow or gray coloration of the fundus under light conditions. After two to three hours in total darkness, the normal color of the fundus returns- known as Mizuo’s phenomenon)
• Fundus albipunctatus. • Fleck retina of Kandori |
|
|
RP visual field:
|
Insidious progressive loss of peripheral VF.
ii. Early: relative scotomas in the mid-periphery, between 30-50° from fixation. iii. Later: scotomas enlarge, deepen and coalesce to form a ring of field loss: a. Gradually enlarge toward the far periphery. b. Islands of relatively normal field can remain until late. iv. Inner margin contracts slowly toward central field, producing tunnel vision. v. Long after entire peripheral field is gone, a small oval of intact central field usually remains |
|
|
Central Vision loss seen in RP
|
3. Central vision loss:
i. Central visual acuity may be preserved until the end stages of RP. ii. Can be affected early by: a. CME. b. Diffuse retinal vascular leakage. c. Macular pre-retinal fibrosis. d. RPE defects. |
|
|
Relation of VA to type of RP:
a. Sector RP: ____________ b. Autosomal dominant RP: ___________. c. Autosomal recessive RP: ________ d. X-linked recessive RP: ___________________ |
a. Good vision throughout life.
b.More likely to retain good vision beyond 60 yrs of age c.Less likely to retain good vision beyond 60 yrs of age. d.VA usually 20/200 or worse (equal to ≤ 20°) by age 30-40. |
|
|
Top causes of flashing lights:
|
1. PVD.
2. Migraine: shimmering zigzag, and move off to side. 3. Deep and subretinal inflammation (i.e. inflammatory white dot syndrome). 4. RP. |
|
|
Early fundus findings of pt with RP
|
1. Early, appears near normal
i. “Retinitis pigmentosa sine pigmentoso”: do not have pigment clumps early. ii. May exist for decades. 2. Attenuated retinal vessels,narrowed. 3. Mottling and granularity of the RPE. 4. Bone-spicule intraretinal pigmentation occurs later in disease: |
|
|
What causes bone spicule appearance in RP?
|
4. Bone-spicule intraretinal pigmentation occurs later in disease:
i. Migration of pigment into the retina from disintegration of RPE cells. ii. Accumulates in the interstitial spaces surrounding retinal vessels, especially at the junctions of vessels. |
|
|
Late fundus findings of RP
|
Atrophy of RPE and choriocapillaris, with visibility of larger choroidal vessels.
Macular changes: a. Drier or smoother b. In some, atrophy (bull’s eye atrophy): iii. CME: a. Pigmentary changes around the macula.Common cause of decreased vision. 7. Optic nerve head pallor: Waxy pallor of the ONH. ii. Late: optic atrophy and/or pre-papillary gliosis. iii. Disc drusen in 10%. on “Coats’-like”: telengiectatic vessels that leak exudates |
|
|
Presence or absence of ___________________ is of significant prognostic importance with regard to retention of visual acuity over the next 5 years in pts with RP
|
macular RPE defects
Absence of macular lesion was associated with only one line of acuity loss over the 5-year period. |
|
|
Most common cause of vision loss in RP?
|
CME!
|
|
|
Refractive status of most RP pts?
|
Increased prevalence of high myopia and astigmatism.
|
|
|
Most reliable method of quantifying change in visual deficit in RP pts?
|
Visual Field--> 2-color static perimetry
|
|
|
Scotopic (rod-dominated) ERG is affected to a much more severe degree than the photopic (cone-dominated) ERG in which type of RP?
|
type I RP.
Early diffuse loss of rod sensitivity relative to cone sensitivity. 2. Childhood-onset nyctalopia. |
|
|
Type I RP:
|
1. Early diffuse loss of rod sensitivity relative to cone sensitivity.
2. Childhood-onset nyctalopia. |
|
|
Type II RP:
|
1. Regional and combined loss of both rod and cone retinal nyctalopia.
2. Adult-onset nyctalopia. 3. More common in simplex disease (sporadic or acquired). |
|
|
In Type 2 RP how is ERG is effected?
|
Scotopic and photopic ERG’s are more equally abnormal in type II RP.
(adult onset nyctalopia) |
|
|
On an ERG, B-wave amplitude deficit is ___________ than a-wave amplitude deficit, both scotopically and photopically, for most forms of RP.
|
relatively greater
|
|
|
What type of RP is it, if there is no family history of anyone else other an your pt who has RP?
|
Simplex:
a. Situation when there is no family history of RP. b. 15-63% of cases (35% of cases in the US). c. Assumed that most of these (up to 70%) represent recessive inheritance. |
|
|
_________RP:
a. Must remain a provisional Dx for 10 years of follow-up to exclude unusual presentations of generalized RP. b. Best prognosis. c. Pigmentary changes are limited to one or two quadrants. d. VF abnormality only in regions of retinal pigmentary degeneration. e. Relatively good ERG responses. |
Sector
|
|
|
In RP inversus (peri-central and central RP): disease mostly confined to the __________.
|
posterior pole
|
|
|
_______________:
i. Retinitis pigmentosa along with autosomal recessive congenital deafness. ii. Accounts for 50% of persons who are both deaf and blind. |
Usher’s syndrome:
|
|
|
Cone-Rod Dystrophy (CRD):
a. General features: |
1. Symptoms relating to the cone dysfunction occur initially and are predominant.
i. No symptoms of cone dysfunction at birth (unlike rod monochromatism) a. Decreased central vision 1. May PRECEDE fundus abnormalities. 2. 20/200 late. b. Photophobia: patients prefer dim illumination. c. Severe color vision defect: usually present only after VA worse than 20/40. Dark adaptometry: Abnormal cone portion and normal rod portion until late. |
|
|
Management/Treatment for Cone-Rod Dystrophy
|
f. Management:
1. No medical treatment is available. 2. Gray-tinted glasses with 20% light transmission and an additional front-surface metallic coating for outdoor use (reduce the transmission of light below the 4% level), helps with photosensitivity |
|
|
Pseudoretinitis pigmentosa often involve Retinal inflammatory diseases such as:
|
a. Rubella retinopathy
b. Syphilis c.IInfectious retinitis d. Cancer-associated retinopathy (CAR): e.Melanoma-associated retinopathy (MAR) f. Drug toxicity |
|
|
Cancer-associated retinopathy (CAR) Symptoms:
|
i. Vision loss: can progress rapidly or slowly.
ii. Halo of missing peripheral vision: ring-like scotoma. iii. Central or paracentral positive visual phenomena: a. “Shimmering” or “dancing” lights. b. Photopsias. iv. Night blindness. ERG severely abnormal( extinguished) or rod responses larger than cone responses. |
|
|
Thioridazine Drugs: Phenothiazines bind to________and concentrate in uveal tract and RPE.
Toxicity Symptoms include: |
melanin
Blurring of central vision, poor night vision, brownish discoloration to vision. a. Large “signal” plaques of pigment. b. Diffuse, patchy atrophy of choriocapillaris, RPE, and overlying retina, simulating choroideremia. c. Patient can go blind |
|
|
Thioridazine Drugs: Chloroquine:
Toxicity usually occurs total dose of: Ocular finding: NOTE: Findings for Hydroxychloroquine (Plaquenil) are similar |
total dose of 300 grams.
Bull’s-eye maculopathy: permanent and will worsen even when drug is stopped. Mid-peripheral pigmentary changes with bone spicules can occur. Do amsler and color test on these patients |
|
|
Traumatic retinopathy: Acquired retinopathy that is most commonly confused with RP!!!
Along with_______, account for many cases of misdiagnosed “unilateral RP”. |
DUSN
|
|
|
many cases of misdiagnosed “unilateral RP" are usually the result of
|
DUSEN or Traumatic Retinopathy
|
|
|
Treatment of macular edema seen in RP includes:
|
Diamox 500 mg/day , then decrease to 250 mg/day: Reduces CME in 50% in pts.
|
|
|
T/F Pts with RP should be exposed to, and invest in therapies such as
a. Light deprivation. b. Therapeutic bee stings. c. Vasodilators. d. Injections of placental tissues. e. Russian treatment: intramuscular or peri-bulbar injections of ENCAD, an RNA extract of yeast. f. Cuban treatment |
FALSE
all are WORTHLESS treatments |