Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
76 Cards in this Set
- Front
- Back
|
Contrast central and peripheral tolerance.
|
CENTRAL
deletion of auto-reactive lymphocutes in primary lymphoid organs PERIPHERAL control of T/B cell activation in secondary lymphoid organs [T cells need co-stimulation, B cells need T cell help] |
|
|
How would a T cell see non-thymic antigens whilst still in the thymus?
|
AIRE
|
|
|
What is involved in peripheral activation of T cells?
|
1. Ag recognition
2. Co-stimulation (CD80, CD86 on DC binds CD28 on T cell) |
|
|
How do T cells develop in the thymus?
|
IN THE CORTEX:
1. Lymphocyte pre-cursor undergoes TcR-rearrangement 2. Double positive T cell exposed to APC (i.e. MHC) 3. Positive selection if recognise MHC to become single positive T cell (i.e. CD4+ or CD8+) MEDULLA 4. Negative selection of single-positive T cells (those that recognise self-peptide are killed) |
|
|
If a CD8+ T cell solely recognises MHC class I, what will happen to it?
|
No co-stimulation means that the cell will undergo ANERGY.
[this needs to happen because MHC-I is expressed on all cells, constantly displaying self-peptide] This is an example of peripheral tolerance |
|
|
What is the role of a regulatory T cell?
|
T regs inhibit auto-reactive T cells.
CD25+ CD4+ Recognise self-peptide and inhibit proliferation of disease-causing activated T cells Develop in thymus or during induction of immune responses in peripheral lymphoid organs |
|
|
What do T regs express?
|
Foxp3 = Transcription factor
|
|
|
What inhibitory cytokines do T regs produce?
|
IL-10
TGF-beta |
|
|
T/F: Everyone acquires autoimmune antibodies as they age
|
TRUE!
But they don't always cause disease (low affinity, not pathogenic, IgM, etc) |
|
|
What "T cell help" does a B cell require to be activated?
|
IL-4
CD40L from T cell (binds CD40 on B cell) |
|
|
How do B cells present to T cells for help? Then what happens?
|
B cell = APC.
1. B cell takes up antigen (binds BCR) 2. Antigen processed (phagolysosomal pathway) and loaded onto MHC Class II 3. B cell presents to T cell 4. T cell provides help (cytokines, CD40L) 5. B cells class switch (higher affinity now) 6. Plasma cells become long lived |
|
|
What controls T cell expansion?
|
Fas-FasL
|
|
|
What are the features of autoimmunity?
|
Precipitated by interaction of environmental factors (eg infections) with genetically susceptible host
Occurs mainly in secondary lymphoid tissue Female predominance Chronic fluttering course (relapse/remission) Occurance with other autoimmune diseases Lymphoid cell infiltration of tissues Presence of autoantibodies Response to anti-inflam/immunosuppressive drugs |
|
|
What genes are associated with rheumatoid arthritis?
|
HLA-DRB1 locus
PTPN22 (normally turns off signalling in T cell and macrophages) Other possible genes = CTLA4, IL-2ReceptorA, genes in the TNF signalling pathway |
|
|
What is the incidence in autoimmune diseases in identical twins?
|
Less than 50%
-->ENVIRONMENTAL FACTORS!!! |
|
|
What is associated with developing anti-cyclic citrullineated peptide (CCP) in RA?
|
Smoking
|
|
|
Organ specific autoimmune diseases are in which "cluster"?
|
Thryo-gastric cluster
|
|
|
Non-organ specific autoimmune diseases are in which "cluster"?
|
Systemic or Lupus cluster
|
|
|
What autoantibodies are found in connective tissue disease?
|
1. Anti-CCP [anti-cyclic citrullinated peptide]
2. ANA [anti-nuclear antibody] -->to various components of the nucleus e.g. anti dsDNA antibody 3. ACA [anti-cardiolipin antibody, or anti-phospholipid antibody] 4. Rheumatoid factor [IgM anti-IgG antibody] |
|
|
What is SLE?
|
Lupus
Ag = dsDNA Immune complex driven process [Immune complexes precipitate in various sites in body - complement activated - C3a inflammation] |
|
|
Outline stages of pathogenesis of autoimmune disease (using RA as example)
|
1. INDUCTION
-->DCs in synovium undergo maturation, migrate to lymph nodes and pick up self-peptide -->Activate CD4+ T cells and then migrate back to tissues 2. PERPETUATION -->Activated CD4+ T cells release cytokines causing inflammation -->Activated CD4+ T cells help B cells to produce Abs e.g. RF, ANA 3. CHRONIC FLUCTUATING PHASE -->Tissue damaged by inflammation -->Dysregulation involving multiple cells, local upregulation of MHC-II causing more damage -->Spreading of autoimmune response, self-reactivity spreads to more antigens |
|
|
What do synovial inflammatory cells release in rheumatoid arthritis ?
Which plays a central role in the inflammatory response |
TNF, IL-1, IL-6, IL-12, + others
[TNF plays central role] |
|
|
What are the principles of diagnosis of an autoimmune disease?
|
1. Confirm presence of disease
2. Categorise disease 3. Determine extent 4. Determine severity |
|
|
What are the principles of therapy for autoimmune disease
|
1. Determine extent
2. Anti-inflammatory drugs for mild/moderate disease 3. Immunosuppressants for severe disease 4. Replacement when target tissue destroyed 5. Removal of unwanted autoantibodies/immune compexes (plasmapheresis) 6. Selective inhibition |
|
|
What percentage of NSAIDs are albumin bound?
What does this mean? |
95%
-->small vol distribution |
|
|
What are some side effects of NSAIDs?
|

|
|
|
What is the MOA of NSAIDs?
|

Inhibit COX (cyclooxygenase), thus inhibiting the production of pro-inflammatory prostaglandins
|
|
|
What are the pharmacological properties of NSAIDs
|
Anti-inflammatory
Analgesic Anti-pyretic Anti-platelet |
|
|
What are some normal physiological roles of prostaglandins?
|
COX-1 --> PGE2 and PGI2 are present in the gastric mucosa (give protection)
PGH2 used to produce Thromboxane A2 (aggregates platelets) Prostaglandins maintain normal renal perfusion |
|
|
What is the role of COX-1?
|
Produces prostanoids that mediate physiological role in:
Gastric mucosa Small/large bowel Kidney Platelets Endothelial cells |
|
|
Where is COX-2 expressed?
|
Constitutively expressed in brain and kidney, but induced at sites of inflammation [produces prostanoids that mediate inflammation, pain and fever]
|
|
|
T/F: Cox-2 inhibitors and traditional NSAIDs have similar effects in the kidney
|
TRUE
|
|
|
Which inhibitor would spare platelet aggregation?
COX-2 COX-1 |
COX-2 inhibitor (because platelets contain COX-1)
|
|
|
T/F: COX-2 inhibitors have reduced efficacy
|
FALSE.
The efficacy of COX-2 inhibitors is essentially the same as that of regular NSAIDs |
|
|
Why is there an increased cardiovascular risk with COX-2 inhibitors in patients with atherosclerosis?
|
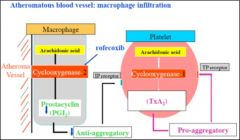
Platelets produce pro-aggregatory TxA2 (because they contain COX-1)
In atherosclerosis, the plaque induces COX-2 (recall induced during inflammation) which synthesises PGI2 (anti-aggregatory). If give selective COX-2 inhibitor, going to inhibit PGI2 production, but not the TxA2 production --> PRO-AGGREGATION (i.e. clotting) [Typical NSAIDs inhibit both PGI2 and TxA2 synthesis] |
|
|
When are corticosteroids clinically indicated
|
Inflammatory disorders (SLE, RA, sarcoidosis etc)
Skin conditions (mainly topical application) Asthma - underlying inflammation Immunosuppression (s.a. for organ transplantation) |
|
|
What are the two major groups of corticosteroids and what are their roles?
|
1. MINERALOCORTICOIDS
(aldosterone) --> maintain electrolyte and water balance (conserve Na, waste K) 2. GLUCOCORTICOIDS (cortisol) -->anti-inflammatory -->glucose, protein, fat metabolism (incr. free glucose, break down protein) |
|
|
Dexamethasone has __ times more glucocorticoid activity than cortisol
|
30
|
|
|
what are the 4 processes identified as the basis for anti-inflammatory activity of glucocorticoids?
|
1. TRANSACTIVATION
synthesis of anti-inflammatory proteins 2. TRANSREPRESSION Switch off inflammatory genes 3. POST-GENOMIC EFFECTS destabilise pro-inflammatory mRNA 4. ACTIVATION OF HDACs [histone de-acetylases] wind up DNA so can't read it |
|
|
How do glucocorticoids mediate transactivation?
|
1. GC travels in plasma bound to cortisol binding protein
2. GC crosses membrane and knocks off heat shock proteins keeping its receptor inactive 3. Active GC-GCR complex goes into nucleus and binds glucocorticoid response element (GRE) on DNA 4. Transcription of mRNA 5. Translation of mRNA into anti-inflammatory proteins (such as annexin-1 which inhibits phospholipase A2) |
|
|
Why may giving corticosteroids cause a better response to beta-agonists in asthma?
|
Glucocorticoids cause translation of proteins that are very similar to beta-receptors in airway smooth muscle. Give asthma a beta-agonist -> will bind to these proteins -> better response because more beta-receptors in ASM
|
|
|
Histone acetylation allows what?
|
Unwinding and thus reading of DNA
|
|
|
How do glucocorticoids mediate transrepression?
|
Aim of transrepression = to switch OFF inflammatory genes.
NF-KB is a pro-inflammatory TF that has intrinsic histone acetylation property (to unwind and read DNA). Corticosteroid-R complex can bind NF-KB, inhibiting its histone acetylation activity End result = repress the reading of genes that would have led to production of pro-inflammatory proteins |
|
|
How do glucocorticoids mediate post-genomic effects?
|
Corticosteroids inhibit stabilising proteins that would otherwise protect pro-inflammatory mRNA
|
|
|
How do glucocorticoids mediate histone de-acetylation?
|
Acetylation of histones causes unwinding of DNA allowing gene transcription and synthesis (of pro-inflammatory proteins)
Histone deacetylases (HDACs) deacetylate acetylated histones -> the DNA remains wound -> GENE SILENCING The GR complex (active receptor + steroid) facilitates the activity of HDACs |
|
|
What is the preferred agent for long term corticosteroid therapy?
|
Prednisone - once/day
|
|
|
How does changing the dose of prednisone change its activity?
|
Low dose = physiological
Medium dose = anti-inflammatory High dose = immunosuppressive |
|
|
T/F: Side effects are reversible on cessation of corticosteroid treatment
|
TRUE
|
|
|
What are some SHORT TERM side effects of taking high dose prednisone?
|
Weight gain
Mood changes Incr. blood glucose Hypokalemia Transitory HPA axis suppression |
|
|
what are some LONG TERM effects of taking corticosteroids?
|
Oedema/weight gain
Infection (immunosuppressed) Osteoporosis Incr. blood glucose Muscle wasting Thin skin |
|
|
How can corticosteroids cause osteoporosis??
|
Cause protein breakdown
Incr. urinary calcium losses Inhibit gut Ca absorption HPA axis suppression -> decr. sex steroids |
|
|
Why is there HPA axis suppression with use of corticosteroids ?
|
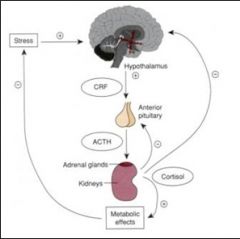
High dose of corticosteroid - feedback inhibition - reduce production of cortisol.
Long term use can completely shut down endogenous cortisol production |
|
|
Excess corticosteroid can lead to what?
|
Cushing's syndrome.
|
|
|
What characterises Cushing's syndrome?
|
Moonface – redistribution of fat
Easy bruising – protein disappears from skin, blood vessels aren’t very well supported [trivial trauma → vessels rupture] Trunk obesity – redistribution of fat Striae/stretch marks – abdominal wall expands, skin is thin, blood vessels rupture Thin limbs – muscle wasting Poor healing – immunosuppression Buffalo hump – redistribution of fat above cervical spine Osteoporosis ↑ BP due to Na retention Thin skin – loss of skin protein |
|
|
Give two examples of using recombinant proteins as drugs.
|
Can use recombinant DNA technology to make proteins directed against other proteins (i.e. antibodies or receptors).
1. Anti-TNF-alpha antibodies can be made and used in RA. These antibodies bind TNF-alpha before it reaches the cell membrane 2. Soluble receptors can be synthesised (e.g. TNF-alpha binds to the receptor thinking it's found the cell, but ends up floating around in the ECF) |
|
|
What is TNF-alpha's role in the inflammatory response?
|
Induces pro-inflammatory cytokines (such as IL-1)
Increases adhesion molecules on epithelial cells Activates neutophils and eosinophils |
|
|
What are the criteria for diagnosing RA?
|
AT LEAST FOUR OF THE FOLLOWING:
1.Morning stiffness > 1hr [6+ wks} 2.Arthritis of ≥ 3 joints [6+ wks} 3.Arthritis of hand joints [6+ wks} 4.Symmetrical arthritis [6+ wks} 5.Rheumatoid nodules 6.Serum rheumatoid factor 7.Typical radiographic changes |
|
|
What is the epidemiology of RA?
|
1% of world’s population
3-5x more common in women 50% slow onset, 25% acute onset Both local and systemic disease MTDx (mean time to diagnosis) = 9 months |
|
|
What are some proposed causes of RA?
|
Triggered by exposure of a genetically suscepible host to an antigen which causes a breakdown in self-tolerance.
Infectious agent in synovium? Continued presence of super antigen? Altered T cell repertoire? Decr. T regs? |
|
|
What ultimately destroys the joint in RA?
|
The CD4+ T cell activation, and local release of inflammatory mediators/cytokines
|
|
|
What cells are in the synovial membrane?
|
Type A synoviocytes (equivalent to macrophages)
Type B synoviocytes (equivalent to fibroblasts - produce synovial fluid) |
|
|
How thick is the synovial lining?
|
One cell
|
|
|
What are the characteristic histological features of ACUTE RA?
|
1. Increased vascular flow
2. Aggregation of organising fibrin (can float in joint space = rice body) 3. Endothelial activation so cells can migrate into tissue 4. vascular proliferation (HEV, angiogenesis) 5. Replication Type B synoviocytes 6. Recruit type A synoviocytes (recall like macrophages) 7. Type A and Type B FUSE --> GIANT CELL 8. Pannus formation (cells are proliferating with nowhere to go so they protrude into the synovial space) |
|
|
Which immune response elements are present in the acute response to RA?
|
Innate = synovial fluid neutrophils, NK cells, macrophages
Adaptive = B1 cells, CD4+ T cells, Soluble recruitment factors |
|
|
What CHRONIC changes are seen in RA?
|
1. Recruitment (macrophages, neutrophils, T/B cells)
2. Remodelling (inflammatory process has done a lot of damage via MMPs, macrophages, ROS, NO .. have TIMPs. 3. Decr. apoptosis of synoviocytes (anti-apoptotic factors s.a. bcl-2) 4. Hypoxia induced proliferation 5. Synoviocyte fusion 6. Pannus formation 7. Cartilage/bone erosion (juxta-articular erosion, subchondral cysts, osteoporosis) |
|
|
What radiological features would you see in RA?
|
Joint narrowing
Erosions adjacent to cartilage Osteopaenia |
|
|
What is the 'pannus' ?
|
Pannus = mass of synovium stroma consisting of inflammatory cells, granulation tissue and synovial fibroblasts which grows over the articular cartilage and causes its erosion.
|
|
|
What is within the 'pannus' ?
|
Hypoxia in Type B synoviocytes --> Incr. HIF-alpha --> makes VEGF --> poorly ordered angiogenesis
Have HEV instead of normal BVs Type A synoviocytes producing IL-1, IL-6, TNF-a, IL-17, IL-23 Type B synoviocytes producing M-CSF, TNF, IL-17, TGF-b MMPs DCs (Continuous antigen) B cells -> Abs -> immune complexes (i.e. RF) |
|
|
What are some genetic polymoprhisms associated with RA?
|
- Genetic susceptibility clearly major contributor to pathogenesis of RA
- Class II MHC – Specific HLA-DRB1 alleles have been shown to be associated with RA - Class II MHC-linked – TNF-α, heat shock protein-70 - Hormone-related genes – prolactin, oestrogen, … - Lymphocyte-associated genes - Cytokines and cytokine receptors |
|
|
What is the function of synovial fluid?
|
Efficient lubricant
Vehicle for nutrients and oxygen to articular cartilage |
|
|
What is the function of articular cartilage?
|
Shock absorber
Smooth, friction-free surface |
|
|
Why is there an irregular junction between cartilage and subchondral bone?
|
To resist shearing forces
|
|
|
What do the ligaments and joint capsule consist of?
|
Dense, collagenous connective tissue.
--> strong and stable junction between bones |
|
|
What is in the matrix of articular cartilage?
|
Hydrophilic glucosaminoglycans trapped in inextensible collagen fibril framework
|
|
|
T/F: articular cartilage lacks a periosteum
|
FALSE. lacks a perichondrium.
o Develops within a continuous tube of developing collagenous connective tissue that becomes periosteum over the surfaces of the adjacent bones |
|
|
Define the immune response in RA in a sentence.
|
T cell driven initiation followed by macrophage dominated perpetuation and joint destruction
|