![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
47 Cards in this Set
- Front
- Back
|
Platelets - general features |
•Highly complex cell “fragments”derived from megakaryocytes. •Circulate at ~150,000 – 450,000 /μlblood •Metabolically active •Critical for thrombus formation |
|
|
Crucial platelet ligands/activators (8) |
|
|
|
Dense granule contents |
-ADP/ATP -Ca2+/Mg2+ -Polyphosphates -Serotonin -Histamine |
|
|
Alpha-granule contents |
•Integral membrane proteins(P-selectin, αIIbβ3, GP1b) •Procoagulants (fV, fIX) •Plasminogen •Adhesion proteins (fibrinogen, vWF) •Chemokines •Growth factors •Immune mediators |
|
|
Platelet function |

|
|
|
Activated platelets + clot formation |
Activated platelets directly participate in clot formation - expression of VIIIa/Va on surface (intrinsic/common pathwya and eventual activation of II to IIa) Secondary hemostasis (clotting cascade) can occur on the surface of activated platelets (amplification of clotting cascade) |
|
|
Manifestations of platelet-related bleeding disorders |
-Easy bruising -Mucosal bleeding: epistaxis, heavy menstrual bleeding, gum bleeding, GI bleeding -Bleeding with surgical challenges can be highly variable |
|
|
Bleeding time
|
Normal <9.0 minutes -Requires skilled technician and cooperative patient -Rather insensitive to vWD (which is bad - this disorder is very common) -Very sensitive to aspirin-like diseases |
|
|
PFA-100 |
Platelets hit with potent agonist while whole blood is flowing through capillary tube; measures time to cessation of flow Agonists: 1) Collagen/epi: primary screening cartilage 2) Collagen/adp: differentiates dysfunction due to aspirin |
|
|
Caveats to PFA-100 |
|
|
|
Thromboelastogram |
Whole-blood clotting assay Whole blood put in cuvette and clotting system activated via TF or contact pathway Torsional stress put on pin as clot is formed + machine measures this torsional stress Variation in measurements - time to start forming clot, maximum amplitude |
|
|
Advantages of thromboelastogram |
|
|
|
Potential disadvantages to thromboelastogram |
-Standardization -Uses whole blood -Relatively unaffected by low dose ASA -Age based differences in normal ranges not defined |
|
|
Platelet aggregometry |
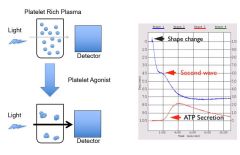
When agonist added - initial shape change Platelets quickly start to aggregate - primary wave Secondary wave of aggregation - after dense granules released - recruit more platelets Measure ATP secretion w/ bioluminescence - measure of platelet granule release |
|
|
How can you assess platelet granule release? |
•Adenine nucleotide content– are dense granules present in normal numbers? •Quinacrine uptake (intodense grandules) andrelease – are dense granules present/released properly? •ATP secretion with aggregation •Platelet electron microscopy-look at granules and count them |
|
|
Glanzman's thrombasthenia - defect, common presentation, platelet behavior |
•Autosomal recessive defect in αIIbβ3 (GP IIb/IIIa), the integrin receptor for fibrin •Purpura and mucocutaneous bleeding •αIIbβ3-deficient platelets display impaired spreading, clot retraction and granule release. |
|
|
Three subtypes of Glanzman's |
–Type I: αIIbβ3 levels < 5% of normal –Type II: αIIbβ3 levels 5 – 15% of normal –Variant disease |
|
|
Diagnosis of Glanzman's thrombasthenia |
•Platelet number, size and morphology arenormal •Platelet aggregometry: αIIbβ3-deficient platelets fail toaggregate in response to all agonists except vWF/ristocetin (used in vitro - binds vWF and allows it to bind platelets statically w/out need for flow) •Flow cytometry •Gene sequencing |
|
|
Bernard-Soulier syndrome - defect, inheritance, proteins involved in affected complex, mutations |
•Defect in the vWFreceptor – GPIb/IX/Vcomplex. •Autosomal recessive •The receptor of a product of 4 distinctgenes – GPIbα,GPIbβ (most often mutated), GPIX and GPV •Mutations resulting in loss of expressionand impaired function have been described. |
|
|
Diagnosis of Bernard-Soulier syndrome |
•Platelet aggregation is normal inresponse to ADP, collagen, TRAP and epinephrine. •Blunted aggregation in response tothrombin. GPIbαcontains high-affinity thrombin binding sites. •Absent platelet aggregation in responseto ristocetin,which is not corrected by normal plasma. •Flow cytometry can also be very useful toquantitate GP Ib/IX/V expression. |
|
|
Platelet-type vWD - mutation, what does it mimic, how do you distinguish these |
•Resultsfrom a gain-of-function mutation in the GPIb/IX/VvWFreceptor •Closelymimics Type 2B vWD. Both exhibit decreased vWF and fVIIIlevels in plasma, loss of large vWF multimers, andincreased sensitivity to ristocetin •Canbe distinguished by mixing studies. Administration of normal vWF towashed platelets causes spontaneous aggregation in platelet-type vWD butnot Type 2B •Receptorand vWFsequencing also available. |
|
|
Rare platelet receptor defects |
•Defects in the collagen, ADP andthromboxane receptors have been described •Abnormal aggregation in response tocollagen usually indicates aspirin use or a storage pool defect. |
|
|
MYH9-Related Macrothrombocytopenia - defect, where is this gene expressed |
•Defects in MYH9 gene (22q12) - non-musclemyosin heavy chain IIA. •MYH9 is expressed in megakaryocytes,macrophages, neutrophils, kidney, eye and cochlea. |
|
|
MYH9-Related Macrothrombocytopenia - presentation triad, other symptoms associated |
•Classic triad of mild thrombocytopenia, Döhle-likeinclusion bodies in granulocytes and giant platelets. •Can be associated with Alport-likesyndrome symptoms: nephritis, deafness, and cataracts. |
|
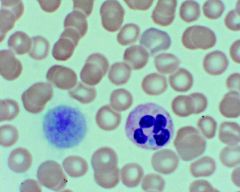
Histo findings? What disease process indicated? |
Giant platelet and Dohle body in the neutrophil - MYH9-Related Macrothrombocytopenia |
|
|
Manifestations of MYH9 Related Macrothrombocytopenia - inheritance, triad, what has variable penetrance, what else contributes to development of non-hematological abnormalities? |
-Autosomal dominant -Classic triad of thrombocytopenia, giant platelets, and Dohle-like bodies in granulocytes -Variable penetrance of nephritis, deafness, and cataracts -Other genetic and environmental factors are likely to be important in the development of the non-hematological abnormalities |
|
|
Diagnosis and management of MYH9-Related Macrothrombocytopenia |
•There is no known treatment or preventionfor the nephritis or hearing loss. •Bleeding manifestations are mild. •Platelet transfusion may be indicatedprior to major surgeries. •Genetic testing in available. |
|
|
Disorders of platelet granule release |
•Heterogeneous group of disorders withvaried clinical manifestations. •Generally characterized by a defect inthe second wave of aggregation after stimulation with ADP and epinephrine. •Primary wave is generally present but maybe impaired. (second wave is abnormal and may even see disaggregation) |
|
|
Platelet storage pool deficiencies |

|
|
|
delta storage pool deficiency manifestations and diagnosis |
|
|
|
alpha storage pool deficiency AKA |
grey platelet syndrome |
|
|
Grey platelet syndrome |
•Autosomalrecessive disorder with platelet-related bleeding and mild thrombocytopenia. •Diminishedα granules in megakaryocytes and platelets •Plateletaggregation responses can vary. •Diagnosisis suspected primarily by visual inspection of Wright stains and EM. •Diminishedplatelet aggregation in response to TRAP •Greyplatelets will express less P-selectin after stimulation than normal plateletsby FACS. |
|
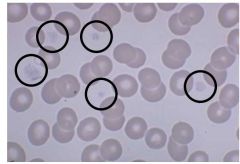
Defect? |
alpha storage pool deficiency - grey platelet syndrome |
|
|
Other abnormalities associated w/ gray platelets |
•Trisomy 21 with MDS •Ongoing platelet consumption leading tostressed megakaryopoiesis. •Dysmegakaryopoiesis |
|
|
Defects in TxA2 secretion/function lead to... |
-Diminished delta-granule release -Mild/moderate bleeding |
|
|
Defects in TxA2 can result from: |
-Abnormal mobilization of arachadonic acid -Defect in COX or TxA2 synthase |
|
|
Aspirin effects |
–Irreversiblyacetylates COX1 –Decreasedaggregation to ADP, epinephrine and collagen –Mildeffect on hemostasis |
|
|
Other NSAIDs effect on platelets |
–Transientimpairment of COX1 –Generallybleeding risk is less than with Aspirin1 |
|
|
Ticlopidine and clopidogrel |
•Diphosphate P2Y12inhibitors: Ticlopidine and clopidogrel(Plavix). Inhibit the binding of ADP toits receptor. |
|
|
Dipyradimole |
•Adenosine Reuptake inhibitors:Dipyridamole. Inhibits thromboxane synthase, prevents reuptake of adenosine.Results in less TxA2 and ADP release. |
|
|
Abcixamab |
•αIIb/β3inhibitors:Inhibit the binding of fibrinogen to its receptor (Abcixamab). VERY POTENT inhibitors of plateletaggregation. |
|
|
Defect in Glanzman's |
Homozygous genetic deletion of the gene encoding the αIIb subunit of the αIIbβ3 integrin receptor |
|
|
What can present similarly to NSAID use? |
Disorders of COX1 or thromboxane synthase will present similarly (but would have more of a bleeding hx) |
|
|
Thepatient is an 11 year old girl from Saudi Arabia with a lifelong history ofsevere mucosal bleeding. In particular, she suffers from daily, severe gingivalbleeding, resulting in a poor diet and failure to thrive. She has chronic irondeficiency anemia due to bleeding. Impaired response to collagen, epinephrine, arachidonic acid, TRAP, ADP induced aggregation. Normal risocetin induced aggregation. Diagnosis? |
Glanzman's thrombasthenia - homozygous genetic deletion of the gene encoding the aIIb subunit of aIIb/B3 integrin receptor |
|
|
Thepatient is a 7 year old who suffered three post-operative bleeding episodesfollowing T&A. Father has a history of significant bleeding followingdental extractions and T&A. Second wave of aggregation is absent or blunted in response to ADP/epinephrine. Decreased quinacrine uptake into platelets. |
Dense granule storage pool disease |
|
|
Thepatient is an 18 year female with heavy menstrual bleeding. No other bleedinghistory. Underwent tonsillectomy and dental extractions without bleeding. Nofamily history of bleeding. Decreased aggregation in response to ADP, epinephrine, collagen; no response to arachidonic induced aggregation. |
NSAID use - disorders of COX1 or thromboxane synthase will also present similarly |
|
|
Treatment of platelet function defects |
•Platelet transfusion – generallyreserved for severe bleeding in the context of Glanzman’s orBernard Soulierbecause of risk of alloimmunization •Local measures – pressure,sealants, cautery etc •Antifibrinolytic therapy – Amicar,Tranexamic acid (particularly for granule storage disease/release defects) •Desmopressin/Humate P {concentration of vonW protein) (desmopressin can be hard to tolerate w/ decreased renal fxn) •NovoSeven(recombinant fVIIa) – particularly helpful for Glanzman’s thrombasthenia |