Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
50 Cards in this Set
- Front
- Back
|
What is the difference between pharmacodynamics and pharmacokinetics?
|
How the drugs changes the body: dynamics
How the body changes the drug: kinetics |
|
|
What is the four step model that encompasses every action of pharmacokinetics?
|
A - Absorption
D - Distribution M - Metabolism E - Excretion |
|
|
inhibitor
|
prevents or reduces a pharmacological activity usually at the level of receptors, cell, organ/tissue, or whole body
|
|
|
activator
|
enhances a pharmacological activity usually at the level of receptors, cell, organ/tissue, or whole body
|
|
|
agonist
|
produces an action at a receptor system
|
|
|
antagonist
|
prevents an agonist from having its action at the same receptor system
|
|
|
full agonist
|
binds to receptor and produces full on molecular response. this allows a full on effect from a normal drug interaction.
|
|
|
Competitive Antagonist
|
binds to the receptor so as to NOT initiate a receptor response
|
|
|
Partial agonist
|
binds to receptor and produces a response, but it's not full on. no matter the concentration of the drug or the partial agonist, maximum will not ever occur. this is competitive relative to the full agonist. snap!
|
|
|
Inverse agonist
|
when this bad boy binds, it cuts down all activity to where NOTHING occurs. there is basal activity going on without any agonist/antagonist action. this cuts the basal activity out so that it is fully zero.
|
|
|
Receptor reserve
|
this is the idea that not all receptors have to be active to be able to achieve the maximum response Emax. In fact the ones that are left over, receptor reserve, are used to detect small changes in concentration
|
|
|
Partial agonism
|
this is when the receptor isn't activated enough to matter, but it is slightly activated. this kinds makes the receptors that are half activated unusable (to produce Emax, anyway)
|
|
|
partial agonism and full agonism dynamic
|
If there are spare receptors, then we can use extra partial agonists and get a full response, but if no spare receptors are available, well... I guess we're SOL.
Likewise, if we have no spares, we can still get a full response from a full agonist, but a weak response from a partial agonist... if both partial and full agonists are available in the same environment, then the partial binds to the receptor first.... oh snap! that partial just became competition, acting as a functional antagonist.... snap! |
|
|
Antagonist
|
reduces the number of receptors available for agonist binding
|
|
|
competitive antagonist
|
fills the hole that the agonist would go in
|
|
|
non-competitive antagonist
|
hits a different hole aka the allosteric site, but causes a conformational change so that the agonist hole is unusable
|
|
|
uncompetitive antagonism
|
like the noncompetitive antagonist, but oh boy! it can bind the allosteric site even when a ligand is bound to the active site. in fact, it can only bind the allosteric site when the agonist activates the receptor.
|
|
|
physiological antagonism
|
special antagonism where there is a physiological event that has an antagonistic effect, like acetylcholine (vaso-dilator) and noradrenalin's (vaso-constrictor) effects on arterioles
|
|
|
What levels can drugs interact with the body?
|
moleular, cellular, tissue/organ, system
|
|
|
What are molecular drug targets?
|
hormone and neurotransmitter receptors, enzymes, transporters, ion channels, nucleic acids, idiosyncratic targets.
|
|
|
What are some drugs that do not have molecular targets?
|
buffers (antacids), gaseous and volatile general anesthetics, osmotic diuretics or laxatives.
|
|
|
What are some prerequisite characteristics of receptors in ligand/receptor interactions?
|
recognition based on:
1) saturability (receptors exist in finite numbers), 2) reversibility, 3) stereoselectivity, 4) agonist specificity, 5) tissue specificity. |
|
|
What happens when the prerequisite characteristics are met for a specific ligand/target interaction?
|
transduction can occur that allows the drug action to take place -> meaningful physiological or biological response
|
|
|
What are the four types of hormonal neurotransmitter receptor superfamilies?
|
1) Receptor-operated channel (ROC) - receptor is activated, transmembrane protein opens channel for transduction
2) Tyrosine kinase receptor (receptor is an enzyme) - also transmembrane, but uses tyrosine kinase catalytic domain to initiate transduction 3) G protein-coupled receptor (GPCR) - like (1) coupled with a G protein 4) DNA-coupled receptor - intracellular receptor that changes conformation of drug, binds, and transfers drug to nucleus to alter translation, transcription |
|
|
What is the lock and key concept?
|
receptors are a specific lock that will only recognize a very specific key (drug). once the lock and key bind a conformational change occurs that allows a physiological effect.
|
|
|
What is Kd?
|
this is the equilibrium constant of the drug, receptor, and drug-receptor complex. Kd = [D] that produces 50% receptor occupancy.
|
|
|
What is the practical meaning of a high Kd?
|
the higher [D] is necessary to achieve 50% occupancy, so the receptor-ligand has a lower affinity
|
|
|
What is the practical meaning of a low Kd?
|
a lesser [D] is necessary to achieve 50% occupancy, so the receptor-ligand has a higher affinity
|
|
|
What is fractional occupancy?
|
the % of receptors at a given time that are occupied by the drug. This value depends on [D] and Kd.
[D]/(Kd+[D]) |
|
|
What is the functional meaning of [D] vs. fractional occupancy on a graph?
|
The larger the Kd, the higher [D] will have to be in order to overcome the Kd value. Consider the equation.
[D]/(Kd+[D]) |
|
|
What happens to the Kd value when there is a competitive inhibitor?
|
The original Kd value is no longer valid, so a Kd' value is defined by [I] and Ki so that
Kd' = Kd*(1+([I]/Ki)) |
|
|
What is the practical change and necessary change to overcome a competitive inhibitor?
|
The practical change is that receptors are occupied by I, so the higher [I] and higher Ki, the more [D] is necessary to overcome and Kd is increased.
On the graph, the curve is simply shifted to the right with a higher [I]/Ki value. You overcome this by increasing [D]. |
|
|
How does the integration of molecular and cellular mechanisms alter the time it takes for certain actions to take place?
|
depends on what the cascade of events is involved. the more complex a cascade is, the longer it will take. example, if a cascade requires transcription, translation, and protein synthesis then it could take hours.
|
|
|
What is a dose-response relationship?
|
This is the effect (response) in the whole human based on the concentration of drug introduced into the system. This is often represented by the Emax model.
|
|
|
What is Emax?
|
The maximum effect an agonist can have.
|
|
|
What is A50?
|
A50 = ED50 = EC50. This is the amount or dose that produces 0.5Emax. This is like the Kd, but for the whole effect of an agonist on the body.
|
|
|
How can we functionally represent Emax on a graph?
|
A system doesn't operate at Emax all the time. It operates at E, so if we graph E/Emax, we can see where A50 is based on E = 0.5Emax.
E/Emax = [A]h/(A50h+[A]h) |
|
|
In general what will E/Emax look like on a graph and why?
|
E/Emax will not be a simple curve or a logarithmic line because on a system level there are phases of signal transduction that involve translation and transcription which needs more drug (and time for that matter). So usually one sees a sigmoidal curve, which makes sense.
|
|
|
What are the three phases of a sigmoidal curve in a log plot of an E/Emax graph?
|
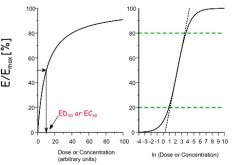
1) linear phase - the slope is linear so that where D << than what's required for Emax, E=mD
2) log-linear phase - for E between 20% and 80% of Emax, the model is log linear to say that on a log plot it is a straight line. 3) constant phase - value is nearly constant with values of E very close to Emax and >> A50, so functionally E = Emax |
|
|
Intrinsic Activity (IA)
|
relative maximum effect of a trug in a particular tissue. this is defined relative to Emax.
Full agonist - IA = 1 Antagonist - IA = 0 Partial agonist - 0 < IA < 1 Inverse aconist - IA < 0 |
|
|
Potency
|
ability of a drug to cause a measured functional change at a certain dose/concentration. characterized by A50. so smaller A50, the more potent a drug is. Easy peasy.
|
|
|
Efficacy
|
defined by receptor occupancy and the ability to initiate a molecular level response
|
|
|
ED50 = A50 = EC50
|
with a graded response, it is what we know, 0.5Emax, but for a binary response, the definition is different:
consider life:death = 1:0 the [D] at which 50% of subjects = 0 is the ED50 in this case, not where each individual gets 50% of Emax. get it? the definition changes a bit. |
|
|
TD50
|
ED50 for undesired, toxic effect. so it's beyond the value of ED50 where a drug gets too big [D] and becomes bad.
|
|
|
LD50
|
dose where 50% of experimental subjects die, so
ED50 < TD50 < LD50 |
|
|
Break down ED50, TD50, LD50
|
ED50 - efficacy of [D] to produces 50%Emax
TD50 - the amount of [D] that hits 50% of toxic E LD50 - the amount of [D] that hits 50% lethal in test tubjects |
|
|
Can an antagonist be used as a drug?
|
heck yes. all different types of competing ligands are used to alter body chemistry to produce a reaction.
|
|
|
What does allosteric inhibition do to the dose/response curve?
|
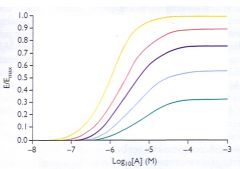
it does not shift the curve to the right the same way that a competitive inhibitor does. Rather, it lowers the Emax (see pic)
|
|
|
What does irreversible inhibition do to the dose/response curve?
|
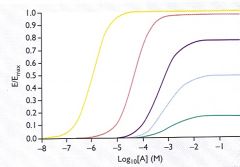
at lower concentrations, it just does a rightward shift because receptor reserve is available. with higher concentrations, as receptor reserve is depleted, it starts to lower Emax (see pic)
|
|
|
How is receptor reserve measured?
|
Kd/A50
|