Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
87 Cards in this Set
- Front
- Back
|
What is the graph of most enzymatic reactions following Michaelis-Menten kinetics? What do the axises represent? |
![Most follow a hyperbolic curve; however, enzymatic reactions that exhibit a sigmoid curve usually indicate cooperative kinetics (eg, hemoglobin)
Y-axis = Velocity (V)
X-axis = [S]](https://images.cram.com/images/upload-flashcards/24/57/23/5245723_m.png)
Most follow a hyperbolic curve; however, enzymatic reactions that exhibit a sigmoid curve usually indicate cooperative kinetics (eg, hemoglobin)
Y-axis = Velocity (V) X-axis = [S] |
|
|
According to the Michaelis-Menten equation, what does Km equal?
|
![Km = [S] at 1/2 Vmax](https://images.cram.com/images/upload-flashcards/24/57/02/5245702_m.png)
Km = [S] at 1/2 Vmax
|
|
|
What is Km (of the Michaelis-Menten equation) related to?
|
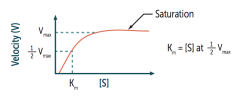
Related to the affinity of the enzyme for its substrate
|
|
|
What is Vmax (of the Michaelis-Menten equation) related to?
|
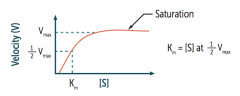
Directly proportional to the enzyme concentration
|
|
|
What is the slope on a Lineweaver-Burk plot? What do the axises represent?
|
![Slope = Km / Vmax
Y-axis = 1/V
X-axis = 1/[S]](https://images.cram.com/images/upload-flashcards/24/57/29/5245729_m.png)
Slope = Km / Vmax
Y-axis = 1/V X-axis = 1/[S] |
|
|
What is the X-intercept on a Lineweaver-Burk plot?
|
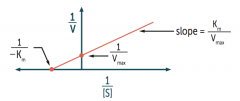
1 / -Km
|
|
|
What is the Y-intercept on a Lineweaver-Burk plot?
|
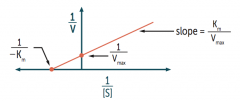
1 / Vmax
|
|
|
What value changes as the Y-intercept increases on a Lineweaver-Burk plot?
|
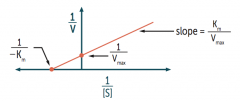
As Y-intercept ↑, Vmax ↓
Remember Y-intercept = 1 / Vmax |
|
|
What value changes as the X-intercept increases (gets closer to zero) on a Lineweaver-Burk plot?
|
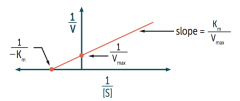
The further to the right the x-intercept, the greater the Km and the lower the affinity
Remember X-intercept = 1 / -Km |
|
|
What happens to the lines of competitive inhibitors on a Lineweaver-Burk plot?
|
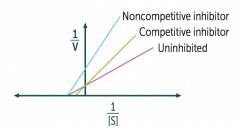
Competitive inhibitors cross each other competitively
|
|
|
What happens to the lines of non-competitive inhibitors on a Lineweaver-Burk plot?
|
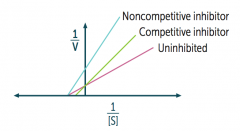
Non-competitive inhibitors do not cross each other
|
|
|
Which types of inhibitors resemble the substrate? Which do not?
|
Resemble substrate:
- Competitive reversible inhibitors - Competitive irreversible inhibitors Do not resemble substrate: - Non-competitive inhibitors |
|
|
Which types of inhibitors can be overcome by ↑ [S]? Which are not?
|
Overcome:
- Competitive reversible inhibitors Not overcome: - Competitive irreversible inhibitors - Non-competitive inhibitors |
|
|
Which types of inhibitors canbind the active site? Which do not?
|
Bind active site:
- Competitive reversible inhibitors - Competitive irreversible inhibitors Do not bind active site: - Non-competitive inhibitors |
|
|
What is the effect of the different inhibitors on Vmax?
|
Unchanged:
- Competitive reversible inhibitors Decreased: - Competitive irreversible inhibitors - Non-competitive inhibitors |
|
|
What is the effect of the different inhibitors on Km?
|
Increased:
- Competitive reversible inhibitors Unchanged: - Competitive irreversible inhibitors - Non-competitive inhibitors |
|
|
What is the pharmacodynamic effect of the different inhibitors?
|
↓ Potency:
- Competitive reversible inhibitors ↓ Efficacy: - Competitive irreversible inhibitors - Non-competitive inhibitors |
|
|
What is pharmacokinetics?
|
The effects of the body on the drug (ADME):
- Absorption - Distribution - Metabolism - Excretion |
|
|
What is pharmacodynamics?
|
The effects of the drug on the body
- Includes concepts of receptor binding, drug efficacy, drug potency, and toxicity |
|
|
Which term encompasses the effects of the body on the drug?
|
Pharmacokinetics, eg:
- Absorption - Distribution - Metabolism - Excretion |
|
|
Which term encompasses the effects of the drug on the body?
|
Pharmacodynamics, eg:
- Concepts of receptor binding, drug efficacy, drug potency, and toxicity |
|
|
What is the term for the fraction of the drug that reaches the systemic circulation unchanged?
|
Bioavailability (F)
|
|
|
What is the bioavailability? What drug/administration has a bioavailability (F) of 100%?
|
Fraction of administered drug that reaches the systemic circulation unchanged
For IV dose, F = 100% |
|
|
What causes decreased bioavailability of orally administered drugs?
|
F typically <100% because of incomplete absorption and first-pass metabolism in the liver
|
|
|
What is the volume of distribution (Vd)?
|
Theoretical volume occupied by the total absorbed drug amount at the plasma concentration
|
|
|
What can affect the apparent Volume of Distribution (Vd)?
|
Apparent Vd of plasma protein-bound drugs can be altered by LIVER and KIDNEY disease (↓ protein binding, ↑ Vd)
|
|
|
How do you calculate the volume of distribution?
|
Vd = amount of drug in the body / plasma drug concentration
|
|
|
What are the compartments in which a drug may distribute?
|
- Blood
- Extracellular Fluid (ECF) - All tissues including fat |
|
|
What compartment is a drug in that has a low Vd? Drug characteristics?
|
Drug is in blood (4-8 L)
- Large / charged molecules - Plasma protein bound drugs |
|
|
What compartment is a drug in that has a medium Vd? Drug characteristics?
|
Drug is in ECF
- Small hydrophilic molecules |
|
|
What compartment is a drug in that has a high Vd? Drug characteristics?
|
Drug is in all tissues, including fat
- Small lipophilic molecules, especially if bound to tissue protein |
|
|
What is the half-life of a drug?
|
The time required to change the amount of drug in the body by 1/2 during elimination (or constant infusion)
|
|
|
A half-life is a property of what type of elimination?
|
First-order elimination
|
|
|
How long does an infused drug infused at a constant rate take to reach a steady state?
|
Takes 4-5 half-lives to reach steady state
|
|
|
How many half-lives does it take to reach 90% of the steady state level?
|
3.3 half-lives
|
|
|
How do you calculate the half-life of a drug?
|
t1/2 = 0.693 * Vd / CL
CL = Clearance Vd = Volume of distribution |
|
|
How much drug is remaining (%) after 1, 2, 3, and 4 half-lives?
|
- 1: 50%
- 2: 25% - 3: 12.5% - 4: 6.25% |
|
|
What does the clearance of a drug represent?
|
The volume of plasma cleared of drug per unit time
|
|
|
What can impair clearance of a drug?
|
Defects in:
- Cardiac function - Hepatic function - Renal function |
|
|
How do you calculate the clearance of a drug?
|
CL = (rate of elimination of drug) / (plasma drug concentration)
CL = Vd * Ke Ke = Elimination Constant |
|
|
How do you calculate what the loading dose should be?
|
Loading dose = (Cp * Vd) / F
Cp = target plasma concentration at steady state Vd = volume of distribution F = bioavailability |
|
|
How do you calculate what the maintenance dose should be?
|
Maintenance dose = (Cp * CL * π) / F
Cp = target plasma concentration at steady state CL = clearance π = dosage interval (time between doses), if not administered continuously F = bioavailability |
|
|
What is Cp?
|
Target plasma concentration at steady state
|
|
|
What happens to the relative maintenance dose in a patient with renal or liver disease?
|
Maintenance dose ↓
|
|
|
What happens to the relative loading dose in a patient with renal or liver disease?
|
Loading dose is unchanged usually
|
|
|
What determines the time to steady state?
|
Depends primarily on t1/2 and is independent of dose and dosing frequency
|
|
|
What are the types of elimination of drugs?
|
- Zero-order elimination
- First-order elimination |
|
|
What type of elimination is characterized by a constant rate of elimination regardless of Cp (constant amount of drug eliminated per unit time)?
|
Zero-Order Elimination
|
|
|
What limits Zero-Order Elimination?
|
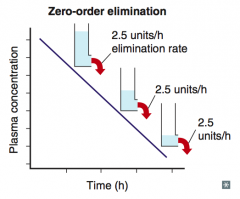
Capacity-limited elimination
(rate is constant no matter what the concentration is) |
|
|
What happens to the concentration of a drug with time with Zero-Order Elimination?
|
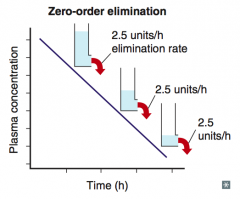
Concentration decreases linearly with time
|
|
|
What drugs have Zero-Order Elimination?
|
PEA (a pea is round, shaped like the "0" in "zero-order")
- Phenytoin - Ethanol - Aspirin (at high or toxic concentration) |
|
|
What type of elimination is characterized by a rate of elimination directly proportional to the drug concentration (ie, constant fraction of drug eliminated per time)?
|
First-Order Elimination
|
|
|
What limits First-Order Elimination?
|
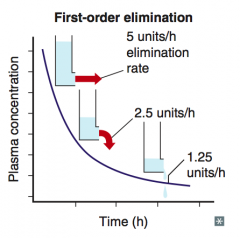
Flow-Dependent Elimination
|
|
|
What happens to the concentration of a drug with time with First-Order Elimination?
|
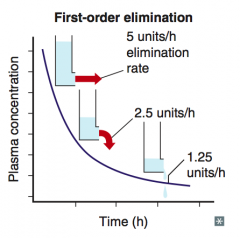
Cp decreases exponentially with time
|
|
|
What gets trapped in urine and is cleared quickly?
|
Ionized species
|
|
|
What drugs are weak acids? How does this affect their elimination?
|
- Ex: phenobarbital, methotrexate, aspirin
- Weak acids are trapped in BASIC environments |
|
|
How do you treat an overdose of a weak acid drug (eg, phenobarbital, methotrexate, aspirin)?
|
Bicarbonate
|
|
|
What drugs are weak bases? How does this affect their elimination?
|
- Ex: amphetamines
- Trapped in ACIDIC environment |
|
|
How do you treat an overdose of a weak base drug (eg, amphetamines)?
|
Treat overdose of weak base with ammonium chloride
|
|
|
What are the phases of drug metabolism?
|
- Phase I
- Phase II |
|
|
What happens in phase I drug metabolism? What does it yield?
|
- Reduction
- Oxidation - Hydrolysis with cytochrome P-450 - Usually yields slightly polar, water-soluble metabolites (often still active) |
|
|
What happens in phase II drug metabolism? What does it yield?
|
- Conjugation (glucuronidation, acetylation, sulfation)
- Usually yields very polar, inactive metabolites (renally excreted) |
|
|
What phase of drug metabolism do geriatric patients lose first?
|
Phase I
- Reduction - Oxidation - Hydrolysis w/ cytochrome P-450 |
|
|
What phase of drug metabolism do geriatric patients usually still have?
|
Phase II
- Conjugation (glucuronidation, acetylation, sulfation) Geriatric patients have GAS |
|
|
What type of abnormal drug metabolism in a patient causes greater side effects from certain drugs? Why?
|
Slow acetylators - decreased rate of metabolism
|
|
|
What is meant by drug "efficacy"?
|
Maximal effect a drug can produce
|
|
|
What is meant by drug "potency"?
|
Amount of drug needed for a given effect
|
|
|
What are some examples of high efficacy drug classes?
|
- Analgesic (pain) meds
- Antibiotics - Antibiotics - Antihistamines - Decongestants |
|
|
What mediates increased potency of a drug?
|
Increased affinity for receptor
|
|
|
What are some examples of high potency drug classes?
|
- Chemotherapeutic (cancer) drugs
- Anti-hypertensive (blood pressure) drugs - Lipid-lowering (cholesterol) drugs |
|
|
What is the effect of a competitive antagonist on potency and efficacy?
|
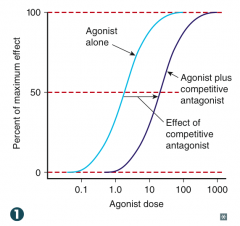
- Shifts curve to right (decreased potency)
- No change in efficacy |
|
|
How can you overcome a competitive antagonist?
|
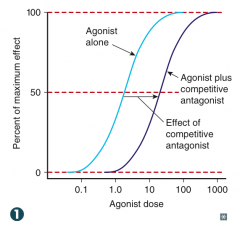
Increase the concentration of agonist substrate
|
|
|
What is the effect of a non-competitive antagonist on potency and efficacy?
|
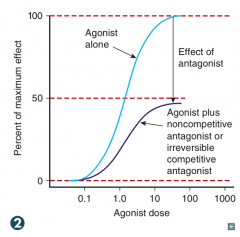
Shifts curve down (decreased efficacy)
|
|
|
How can you overcome a non-competitive antagonist?
|
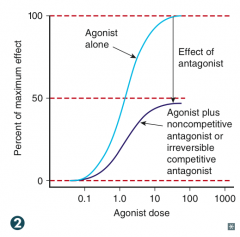
Cannot be overcome by increasing agonist substrate concentration
|
|
|
What is the effect of an irreversible antagonist on potency and efficacy?
|
Shifts curve down (decreased efficacy)
|
|
|
How can you overcome an irreversible antagonist?
|
Cannot be overcome by increasing agonist substrate concentration
|
|
|
What is the effect of a partial agonist on potency and efficacy?
|
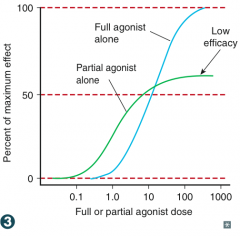
- Acts at same site as full agonist, but with lower maximal effect (decreased efficacy)
- Potency is an independent variable |
|
|
What is the relationship of flumazenil to diazepam?
|
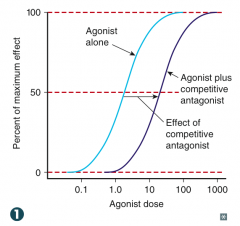
Competitive antagonist at GABA receptor
|
|
|
What is the relationship of ketamine to glutamate?
|
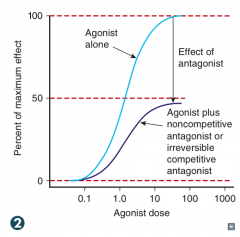
Non-competitive antagonist at NMDA receptor
|
|
|
What is the relationship of phenoxybenzamine to norepinephrine?
|
Irreversible competitive antagonist on α receptors
|
|
|
What is the relationship of buprenorphine on morphine
|
Partial agonist at opioid µ receptors
|
|
|
What measurement is used to assess drug safety?
|
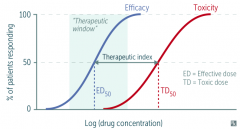
Therapeutic Index
|
|
|
How do you calculate the therapeutic index (TI)?
|
TI = (TD50 / ED50) = (median toxic dose) / (median effective dose)
TITE: Therapeutic Index = TD50 / ED50 |
|
|
What is the therapeutic window?
|
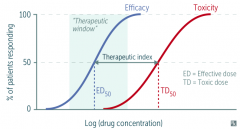
Measure of clinical drug effectiveness for a patient
|
|
|
What relative Therapeutic Index value do safer drugs have?
|
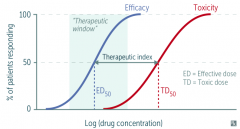
Higher Therapeutic Index values
|
|
|
What are some drugs with low Therapeutic Index values?
|
- Digoxin
- Lithium - Theophylline - Warfarin |
|
|
What value often replaces TD50 in animal studies?
|
LD50 (lethal median dose)
|