Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
83 Cards in this Set
- Front
- Back
|
Which Carbohydrates can be absorbed in the gut?
|
only monosacc.
- the rest can be absorbed by colonic bacteria |
|
|
Amylase
|
Amylase is an enzyme that catalyses the breakdown of starch into sugars.
- found in saliva and pancreas |
|
|
Amylase only targets which bonds?
|
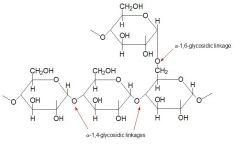
All amylases are glycoside hydrolases and act on α-1,4-glycosidic bonds.
Targets alpha 1,4 glucosidic bonds except when they are terminal or adjacent to an alpha 1,6 glucosidic bond. |
|
|
Main function of salivary amylase
|
Salivary amylase functions as a protection mechanism for teeth by breaking down sugars thereby reducing their availablility for bacterial use.
|
|
|
Brush border membrane enzymes
|
produced by the villi cells and attach to the apical membrane on the luminal surface. They are important in the digestion of di/trisaccharides and peptides
|
|
|
Brush Border Carbohydrate enzymes
|
Sucrase -Isomaltase (breaks down alpha 1,6 bonds of alpha limit dextrins) Lactase (targets lactose); lactase is at the very tip of the villi thus the first to go and last to recover in a GIT illness Glucoamylase |
|
|
Monosaccharide absorption mainly
|
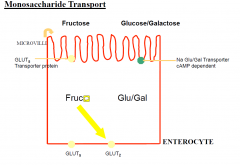
bulk occurs in the top 3rd of jejunum and duodenum
|
|
|
3 key transporters of Monosacc
|
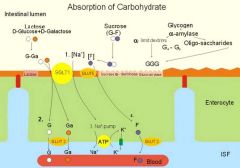
1. Na+/Glucose cotransporter (SGLT1). ACTIVE
□It will also transport galactose and mannose. □Blocked by phlorizin. PASSIVE: 2. GLUT5 will transport fructose and glucose. 3. GLUT2 lies on the interstitial side of the membrane which then transports glucose/fructose to the interstitium. |
|
|
Pepsin produced by
|
gastric chief cells (as zymogen pepsinogen activated by low pH).
|
|
|
Zymogen
|
A zymogen (or proenzyme) is an inactive enzyme precursor.
|
|
|
Pepsin Action
|
Pepsin breaks down protein into AA that act to signal various digestive mechanisms.
|
|
|
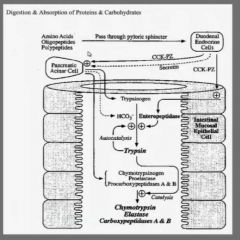
Pancreatic Proteases
|
Pancreatic proteases (all produced as inactive zymogens).
1. Trypsinogen -[enteropeptidase]-> Trypsin 2. Chymotrypsinogen -[trypsin]-> Chymotrypsin 3. Proelastase -[trypsin]-> Elastase |
|
|
Endoproteases
|
Endoproteases cleave various peptide bonds within a polypeptide: Trypsin, chymotrypsin, elastase, kallikrein
|
|
|
Exoproteases
|
Exoproteases start at the end of the polypeptide: carboxypeptidase A and B
|
|
|
Main difference between the abs of CHO and CHN is...
|
that CHN can be absorbed to a certain extent as peptides as well as amino acids
|
|
|
Brush Border Protein Enzymes
|
also found in the cytossol side of the lumen to digest the peptide more
Peptidases Aminopeptidases |
|
|
Absorption of Protein via
|
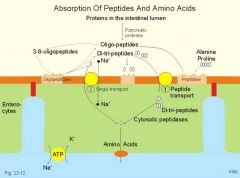
1. PEPT1; H+/protein which is then broken down into a.a through peptidase in the cell
2. System B is a Na+/AA cotransporter that absorbs neutral amino acids into the cell. 3. b0+ -> rBAT/b0+AT (chaperone)/transporter) This transporter is mutated in Hartnup's disease -> An autosomal-recessive disorder characterized by defective neutral amino acid transporter on renal and intestinal epithelial cells. Causes tryptophan excretion in urine and decreased absorption from the gut. Leads to pellagra like dermatoses. |
|
|
Lipases Found in..
|
Saliva
Pharynx Gastric mucosa Pancreas |
|
|
Pancreatic Lipase
|

- TGs broken down by oancreatic lipase to 2 FA + 2-monoacylglycerol
Pancreatic lipase is extremely pH sensitive. It will not function below pH 6. It also needs colipase which is also secreted by the pancreas as procolipase. It facilitates the binding of pancreatic lipase to the lipid globules. |
|
|
Bile Salts
|
○Bile Salts: detergents that increase the surface area for lipases to attach.
- forming micelles |
|
|
Absorption process of Lipids
|
![1. CD36 receptor
2. FA + 2 monoacylglycerol -[intestinal cell]-> Triglycerol
3. Phospholipase A2: release fatty acids from the second carbon group of glycerol.
4. Cholesterol esterase
5. Intestinal cells then package as a chylomicron which is ...](https://images.cram.com/images/upload-flashcards/58/39/40/2583940_m.jpg)
1. CD36 receptor
2. FA + 2 monoacylglycerol -[intestinal cell]-> Triglycerol 3. Phospholipase A2: release fatty acids from the second carbon group of glycerol. 4. Cholesterol esterase 5. Intestinal cells then package as a chylomicron which is secreted into the interstitium to be absorbed by lymphatics. |
|
|
Macronutrients in the diet
|
Protein 20%
Fat 40% CHO 40% |
|
|
Inhested Fats
|
• Triglyceride
• Phospholipids • Cholesterol >95% of ingested fat is triglyceride: cooking oils, margarine, butter, lard, animal fats |
|
|
Triglycerides
|
• Long chain ≥ C14
• Medium chain > C6-C12 • Short chain >C4-C6 |
|
|
Colipase
|
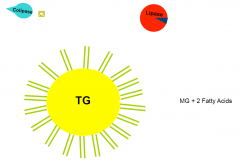
lipase is repeled by bile acids forming around the TGs thus colipase interdigitas b/w them allowing attraction
|
|
|
Mixed Micelle
|
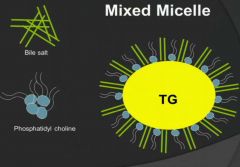
inhibits both colipase and lipase so need to get rid of the phospholipids first
- PLA2 removes the phospholipids, hydrolysed to lysoPC |
|
|
Fats metabolism + absorption
|
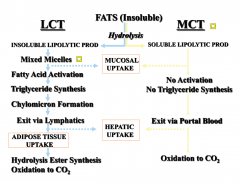
|
|
|
Definition of Fat Malabsorption
|
In a 3 to 5 day fat balance study,
measured faecal fat is >7% of fat ingested |
|
|
Mechanisms of Fat Malabsorption
|
• Maldigestion of ingested fat:
• pancreatic insufficiency • Inadequate micellar solubilisation of digested fat: • bile acid deficiency, as in cholestatic liver disease • Impaired mucosal absorption of fat: • mucosal disease, eg coeliac disease - impaired fat transfer from enterocytes to lymphatics ; lymphatic disease; lymphoma, lympangiectasia - inherited disease; abetalipoproteinaemia |
|
|
Maldigestion of Fat
Pancreatic disease with Pancreatic Insufficiency (PI) |
• PI occurs when >98% of exocrine
pancreas is destroyed or removed • In this circumstance, fat appears in stool as undigested triglyceride, ie oil • In the childhood disorder Cystic Fibrosis, 85% of patients have PI • Approximately 66% of the patients will demonstrate faecal fats between 30 to 50% of fat intake |
|
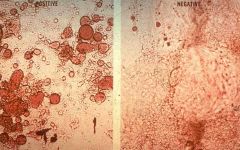
|
Fat droplets in stool
- PI until proven otherwise |
|

|
marked faecal fat (stool from a baby with biliary atresis; a completely blocked bile duct)
|
|
|
Impaired Micelle Formation
|
Usually seen in cholestatic liver disease,
eg bile duct obstruction Cause: Impaired bile acid output into the duodenum • Subsequent low concentration of bile acids below the CMC (critical micellar concentration), approximately 3 mmol/L • Low bile acid concentration • Impaired micelle formation and poor solubilisation of fatty acids and monoglyceride → fat malabsorption – faecal fat 30 to 40% of fat intake |
|
|
Impaired Mucosal Absorption
|
eg Coeliac disease
• Fat malabsorption is quite variable, from 10% to 40% of fat intake • Microscopically, fatty acid crystals (NOT FAT DROPLETS) are found in the stool: Digestion is normal, but discased small intestine fails to absorb fatty acids and monoglycerides |
|
|
Lymphangiectasia
|
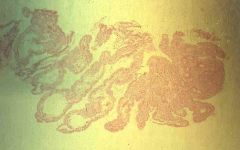
Lymphangiectasia
|
|
|
Abetalipoproteinaemia
|
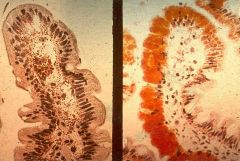
cant get out because of Apopprotein B isnt around
|
|
|
Major Consequences of
Fat Malabsorption |
1. Intestinal
2. Systemic 3. Nutritional |
|
|
1. Intestinal Consequences of fat malabsorption
|
• Diarrhoea
– fatty acids causing fluid secretion – bile acids causing fluid secretion • Bulky, malodorous stools – colonic dilatation • Colonic fermentation – flatulence, gaseous distension of colon |
|
|
Systemic Consequences of fat malabsorption
|
• Bile acid malabsorption
• Interruption of bile acid reabsorption at terminal ileum • Interruption of enterohepatic circulation • Decreased bile acid pool size • Decreased secretion of bile acid formation of lithogenic bile & gallstones |
|
|
Nutritional Consequences of fat malabsorption
|
• Fat malabsorption
– Energy loss, wasting, stunting (long-term) • Fat soluble vitamin malabsorption – Vitamin A D E K deficiencies |
|
|
Vitamin A deficiency
|
• Ocular:
– Impaired dark adaptation (night blindness) – Xerophthalmia (conjunctival dryness) *Bitots spots (small triangular patches in sclera lateral to cornea) – Corneal dryness • Dermatological: - Follicular hyperkeratosis • Neuro: - Benign intracranial hypertension (bulging fontanelle in infancy) |
|
|
Vitamin E deficiency
|
• Premature Infants:
– haemolytic anaemia • Older Children: – progressive neurologic syndrome – areflexia – ataxia – paresis of gaze – spinocerebellar degeneration |
|
|
Vitamin D deficiency
|
Children: Rickets
Adults: Osteomalacia |
|
|
Vit K Def.
|
Coagulopathy
|
|
|
Causes of Protein Maldigestion
/Malabsorption |
• Maldigestion
• Malabsorption • Pancreatic insufficiency – Cystic fibrosis – Chronic pancreatitis – Tumours – Resection • Mucosal disease – Coeliac disease – Short gut syndrome • Protein losing enteropathies • Primary (gut related): – Hypertrophic gastritis (Menetriére’s disease) – H.Pylori gastritis – Inherited lymphangiectasia – Coeliac disease – Inflammatory bowel disease, including eosinophilic gastroenteritis • Secondary: – Cardiac (Constrictive pericarditis, TB, autoimmune) – Liver diseases – Lymphatic obstruction, lymphoma |
|
|
Consequences of Gut Protein Loss
ACUTE |
• Acute
Hypoproteinaemia – loss of oncotic pressure, subsequent loss of fluid to extracellular fluid space with occurrence of oedema, ascites and pleural effusions |
|
|
Consequences of Gut Protein Loss
• Chronic |
– Adults – decrease of lean body mass with
occurrence of muscle wasting (shoulder, girdle and buttock wasting) and (if total body protein drops below 60% of normal) cardiac muscle wasting and cardiac failure – Children – Impaired protein deposition and linear growth failure, noting the most common cause of linear growth failure worldwide is chronic undernutrition / starvation |
|
|
Disaccharide Digestion
|
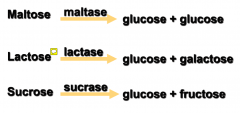
|
|
|
Consequences of Carbohydrate
Malabsorption |
• Failure to digest and/or absorb simple
carbohydrates → osmotic diarrhoea • Colonic fermentation of unabsorbed carbohydrate, production of H2, CH4, gaseous distension and flatulence • Colonic production of short chain fatty acids and colonic secretion of fluid |
|
|
Osmotic Diarrhoea vs Secretory Diarrhoea
|
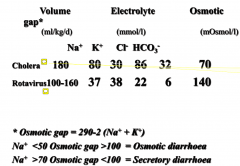
|
|
|
Carbohydrate Malabsorption Inherited
|
• glucose-galactose malabsorption
• lactase deficiency • sucrase-isomaltase deficiency |
|
|
Carbohydrate Malabsorption Acquired
|
• glucose-galactose malabsorption
• disaccharidase deficiencies (severe enteropathies) • excessive intake of fructose and alcoholic sugars (sorbitol & mannitol) |
|
|
Marasmus
|
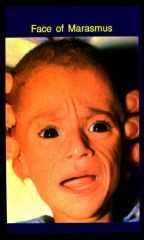
•Protein, energy, vitamin, mineral deficiency
•infancy (weaning or decreased breast milk) •severe weight loss (<60% wt for age) •stunting of linear growth •muscle wasting (when food(energy) intake is insufficient, somatic(muscle) protein is used for gluconeogenesis) •wasting of fat, subcutaneous tissue •oedema not characteristic •Irritable, hyper-alert, ravenous, aged appearance |
|
|
Kwashiorkor
|
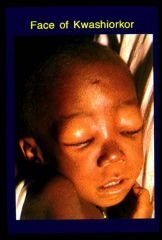
•Pure protein deficiency: inadequate intake &/or increased susceptibility to infection
•older infants and young children •Oedema (decreased serum albumen: decreased protein intake/production (fatty liver) •protein depletion in viscera (liver, gut) •skin (pigmentation, scaling, peeling) •hair (thin, dull, sparse, unnaturally blond) •apathetic, anorexia |
|
|
Kwashiorkor vs Marasmus
|
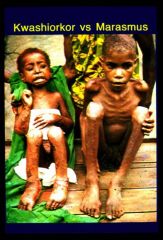
1. Marasmus patients suffer from a peeling and alternately pigmented skin. Kwashiorkor patients are characterized by a distended stomach, burns on the skin and diarrhea.
2. Marasmus affects kids because of a lack of nutritional elements in the diet. Kwashiorkor affects kids who do not receive enough protein in the diet. 3. Marasmus affects infants and very young kids. Kwashiorkor affects kids who are a bit older. 4. Marasmus patients need to be treated with additional doses of vitamin B and a nutritious diet. Kwashiorkor patients are treated by adding more protein in their diet |
|
|
What is malnutrition?
|
Insufficient absorption of nutrients essential for growth and development
•inadequate amount or poor quality food •abnormal absorption of nutrients •increased losses from gut (diarrhoea) •increased energy utilisation eg chronic disease •chronic or recurrent infection eg AIDS Young children more vulnerable •increased growth rate, higher caloric requirements |
|
|
Burden of malnutrition?
|
PEM affects 1 in 4 children worldwide
•27% (150 million) children underweight •33% (182 million) children stunted •70% live in Asia, 26% Africa, 4% Latin America •a spectrum including deficits of protein, energy, vitamins and minerals |
|
|
Stunted
|
•Stunted: Stunted growth refers to low height-for-age, when a child is short for his/her age but not necessarily thin.
•Also known as chronic malnutrition, this carries long-term developmental risks. |
|
|
Under-weight
|
•Under-weight: Under-weight refers to low weight-for-age, when a child can be either thin or short for his/her age.
•This reflects a combination of chronic and acute malnutrition. •Stunted and Under-weight children are most likely to suffer from impaired development and are more vulnerable to disease and illness. |
|
|
Wasted
|
• Wasted: Wasted refers to low weight-for-height where a child is thin for his/her height but not necessarily short. Also known as acute malnutrition, this carries an immediate increased risk of morbidity and mortality. Wasted children have a 5-20 times higher risk of dying from common diseases like diarrhoea or pneumonia than normally nourished children.
|
|
|
Re-feeding Syndrome
|
Any individual who has had negligible nutrient intake for more than 5 consecutive days is at risk of refeeding syndrome. Refeeding syndrome usually occurs within four days of starting to feed. Patients can develop fluid and electrolyte disorders, especially hypophosphatemia, along with neurologic, pulmonary, cardiac, neuromuscular, and hematologic complications.
|
|
|
Burden of diarrhoeal disease
|
•Diarrhoeal disease is the second leading cause of death in children under five years old. It is both preventable and treatable.
•Diarrhoeal disease kills 1.5 million children every year. •Globally, there are about two billion cases of diarrhoeal disease every year. •Diarrhoeal disease mainly affects children under two years old. •Diarrhoea is a leading cause of malnutrition in children under five years old. |
|
|
How are diarrhoea and malabsorption interrelated?
|
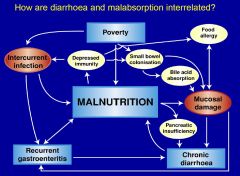
•
Decreased food intake anorexia, pain, therapy • Decreased absorption mucosal damage, motility, osmotic disaccharidases abnormality • Increased energy utilisation (infection) • Recurrent episodes: failure of catch-up • Vulnerability to other infections eg measles •Chronic diarrhoea: >14 days: •reinfection, • ongoing losses: • lactose glucose intolerance, •antigen entry: food intolerance |
|
|
Breaking the cycle
- measures to prevent diarrhoea |
•
Key measures to prevent diarrhoea include: • access to safe drinking-water • improved sanitation • exclusive breastfeeding for the first six months of life • good personal and food hygiene • health education about how infections spread • rotavirus vaccination. |
|
|
Breaking the cycle
- appropriate Tx |
Appropriate treatment
• glucose-electrolyte solution (oral, nasogastric) • Zinc and vitamin A supplementation in the malnourished child • food-based OES • early re-introduction of normal diet • minimise drugs • antibiotics, antiemetics, anti-diarrheals • Management of complications • lactose intolerance • CMPI • recognition re-infection |
|
|
Breaking the cycle: Malnutrition and chronic diarrhoea
6 Rs |
•
Recognise • Resuscitate • Re-feed: • nutrient rich food, breast feeding during an episode • Exclusive breast feeding for first 6 months of life • Replace specific vitamin and mineral deficiencies • Rehabilitate • Rx intercurrent infection |
|
|
Fat Digestion
|
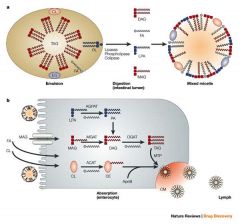
•
Hydolysis by gastric acid • Bile salts solubilise the fat • Pancreatic enzymes such as lipase and colipase digest the fat • Intact intestinal mucosa is required for absorption • Diffusion across enterocyte apical membrane • Reconstitution into chylomicrons with carrier proteins such as apolipoproteinB • Transport via the lymphatic system into the systemic circulation • Small chain triglycerides can bypass this system and enter the portal venous system directly |
|
|
Steatorrhoea (fatty stools)
- aetiology |
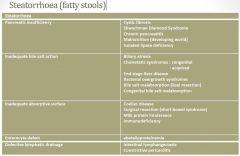
•
Conditions that cause steatorrhoea can also be associated with protein maldigestion and/or malabsorption • symptoms most commonly relate to the malabsorption of fat. • The presence of fat in the stool is also more readily observed than protein. • Diseases of the pancreas and small intestine are the usual causes of steatorrhoea in childhood |
|
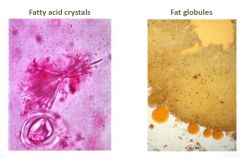
Assessment of fatty stools
|
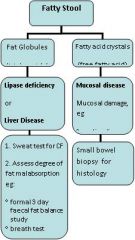
- crystals means brokendown but not absorbed
- and globules = not digested |
|
|
Pancreatic Disease: Cystic Fibrosis
|
•
1:2,500 Caucasians • Chromosome 7, ΔF508 mutation • Meconium ileus • Bilious vomiting newborn • Chronic diarrhoea • pancreatic insufficient • Failure to thirive • Fat soluble vitamin deficiencies (A,D,E,K) • Respiratory illness |
|
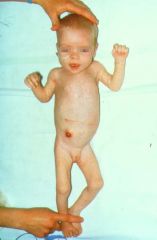
|
This child presented with bile stained vomiting due to meconium ileus (secondary to cystic fibrosis) has a temporary ileostomy. She has abdominal distension, a transverse scar and increased skin folds in the inner thigh suggesting poor nutrition.
|
|
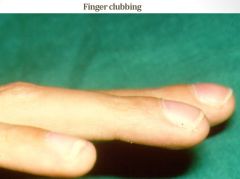
|
Clubbing is characteristic of cystic fibrosis but is also seen in a number of other gastro-intestinal disorders including chronic inflammatory bowel disease (particularly Crohn’s disease), coeliac disease and biliary cirrhosis.
|
|
|
Steatorrhoea in chronic liver disease
|
•
Extrahepatic biliary atresia (other causes neonatal obstructive jaundice) • Cirrhosis Decreased intraluminal bile salts - impaired fat digestion. |
|
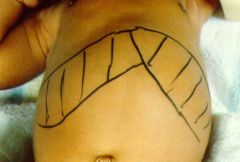
|
This jaundiced child aged 4 months, has marked hepatosplenomegaly, umbilical herniation and abdominal distension.
The width of the liver and spleen from the lower costal margin at the mid-clavicular point should be documented. The liver span can be determined by percussion of the upper border. Liver span varies with age and tables giving normal values are available. Consistency of enlarged organs should also be noted. In this case, the child has extra-hepatic biliary atresia with biliary cirrhosis and secondary portal hypertension and the liver was firm. In children, temporary enlargement of the liver and spleen with viral infections is not uncommon. |
|
|
Coeliac disease
Who should be tested? |
•
Persistent diarrhoea • Failure to thrive • Consider screening in persistent • Abdominal pain • Vomiting • Constipation • Abdominal distension • Irritability * Exclude infection e.g. giardiasis |
|
|
Coeliac disease
From 3 years, even if asymptomatic, |
•Delayed puberty
•Dermatitis herpetiformis •Enamel defects permanent teeth •Osteoporosis •Type 1 diabetes •Short stature |
|
|
Coeliac disease
Regular Screening for |
•Iron deficiency anaemia (Fe resistant or unexplained)
•IgA deficiency, (screening test may be difficult to interpret) •Down syndrome •Turner syndrome •Williams syndrome •1o relatives with CD |
|
|
Coeliac disease: treatment
Gluten free diet for life |
•
Reverses GI symptoms • Maximises growth • Improve bone mineralization • Behaviour change Adults • Decreased intestinal cancers • Decreases spontaneous abortions, LBW infants • Lowers mortality to general population rate ? Gluten dose threshold for harm |
|
|
Carbohydrate Digestion
|
•
Dietary carbohydrates are primarily starch • Starch is comprised of long chains of glucose • Acted on by salivary and pancreatic amylase to trisaccharides, disaccharides, and oligosaccharides • Brush border enzymes disaccaridases break these down to monosaccharides • Sucrase breaks sucrose down to glucose and fructose • Lactase breaks down lactose into glucose and galactose • Glucose and galactose are absorbed by the enterocyte sodium–glucose cotransporter (SGLT), which absorbs the monosaccharides in an energy dependent fashion • Fructose is absorbed by facilitated diffusion (non-energy-dependent) by the transporter termed GLUT-5. |
|
|
Disaccharide Malabsorption
|
•
Causes of disaccharidase deficiencies include: • viral gastroenteritis • coeliac disease • chronic giardiasis • milk protein enteropathy • small bowel bacterial overgrowth syndrome • immunodeficiency disorders • autoimmune enteropathy. |
|
|
Monosaccharide malabsorption
|
•
Monosaccharide transporters are less susceptible to injury because, they are deeply embedded in the brush border membrane • Severe enteropathies can occasionally result in monosaccharide malabsorption. Examples include: • congential villous atrophy (which presents in newborns) • severe postinfectious enteritis • milk protein intolerance • autoimmune enteropathy. • Monosaccharide malabsorption is life-threatening • requires tertiary paediatric level care • treatment is to remove the offending carbohydrate from the diet • substitute an alternative. • In acquired disorders, treatment is required for the primary mucosal disease. |
|
|
Congenital monosaccharide malabsorption
|
•
Congenital monosaccharide malabsorption refers to defective glucose/galactose malabsorption. Features are: • mutations in SGLT1 • recessively inherited • present in the neonatal period. • Treatment is substitution to fructose for glucose–galactose |
|
|
glucose/galactose malabsorption
|
Glucose-galactose malabsorption is a condition in which the cells lining the intestine cannot take in the sugars glucose and galactose, which prevents proper digestion of these molecules and larger molecules made from them.
Glucose and galactose are called simple sugars, or monosaccharides. Sucrose and lactose are called disaccharides because they are made from two simple sugars, and are broken down into these simple sugars during digestion. Sucrose is broken down into glucose and another simple sugar called fructose, and lactose is broken down into glucose and galactose. As a result, lactose, sucrose and other compounds made from carbohydrates cannot be digested by individuals with glucose-galactose malabsorption. |