Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
438 Cards in this Set
- Front
- Back
|
Define the eukaryotic cell cycle
|
Phase G1: Cell growth, p53 error check; S: Synthesis; G2: growth and mismatch repair; Mitosis
|
|
|
Mention Chargaff's rules
|
Strands are antiparallel, complementary and the total % of purines = total % of pirimidines
|
|
|
Mechanism of action: Daunorubicin, doxorubicin
|
Intercalate between bases of DNA interfering with activity of topoisomerase II (DNA gyrase), preventing replication
|
|
|
What is hybridization; Describe denaturing Vs. renaturing
|
Heat, UV light and chemicals are used to denature DNA. Hybridization is when a probe DNA binds to denatured target DNA sequences of sufficient complimentarity.
|
|
|
Function of topoisomerases in prokaryotes
|
Introduce negative or positive supercoiling by breaking and resealing the sugar-phosphate backbone. DNA topoisomerase I (positive supercoiling); DNA topoisomerase II (gyrase) (negative supercoiling)
|
|
|
Define: Histones and nucleosomes
|
Histones are rich in lysine and arginine which confers them positivity; Histones form octamers: H2A, H2B, H3, H4; Nucleosome is DNA + histone octamers linked by Histone 1 (H1)
|
|
|
Euchromatin Vs. Heterochromatin
|
euchromatin = loose, active; heterochromatin = condensed, inactive
|
|
|
What are chromatin-modifying activities
|
Histone acetylation, phosphorilation lessens their positive charge and decreases affinity for DNA
|
|
|
Define: polymerase; exonuclease; endonuclease
|
Polymerase: form phosphodiester bonds to synthesize nucleic acids; Exonuclease: remove nucleotides from 5' or 3' ends; Endonuclease: cut within nucleic acid and release nucleosides
|
|
|
What is an RNA primer?
|
Sequence required for DNA synthesis. Has 5'-3' direction.
|
|
|
Mention the steps of DNA replication in prokaryotes
|
1. DNA A protein recognizes origin sequence; 2. Helicase unwinds by breaking H bonds; 3. SSB prevents reassociation of loose strands; 4. Primase synthesizes RNA primer; 5. DNA pol III synthesizes DNA in 5'-3' direction (Okazaki fragments); 6. DNA pol I removes primers; 7. DNA ligase seals Okazaki fragments; 8. DNA Gyrase (topo II) inserts positive supercoiling.
|
|
|
Define : Reverse transcriptase and AZT
|
It's an RNA dependant DNA polymerase. AZT might be used as a substrate and gets inserted at the 3' end, terminating replication
|
|
|
DNA repair: Thymine dimers
|
1. UV light creates thymine dimers during G1; 2. Removed by excision endonuclease which is deficient in XP; 3. Patched by DNA polymerase and ligase.
|
|
|
DNA repair: Mismatch repair
|
1. During G2 phase; 2. Produced by mutations of MSH2 & MLH1 genes; 3. Associated with HNPCRC
|
|
|
DNA repair: p53
|
Prevents cell with damaged DNA to enter S phase
|
|
|
ATM Gene
|
Encodes kinase essential for p53 activity. Inactivated in Ataxia-Telangiectasia
|
|
|
What are the 6 types of RNA
|
1. rRNA: structural component of ribosome (most common); 2. tRNA: carries amino acids to ribosome; 3. mRNA: contain info for AA sequence; 4. hnRNA precursor of mRNA; 5. snRNA: splices mRNA; 6. Ribozymes: RNA molecules w/enzymatic activity
|
|
|
Define: promoter region
|
The binding site for RNA polymerase. AT-rich sequence with TATA and CAAT boxes
|
|
|
Define: antitemplate (coding) strand Vs. Template strand
|
The antitemplate is not used during transcription but its identical to RNA molecule, except that RNA has U instead of T. RNA polymerase uses template strand 3'-5' to create antiparallel and complimentary 5'-3' strand
|
|
|
What is the direction of translation of the ribosome?
|
reads mRNA 5'-3'; Amino to Carboxyl group.
|
|
|
What is the direction of transcription?
|
reads template strand 3'-5'. makes mRNA 5'-3'
|
|
|
What is the sigma factor; what is the rho factor
|
required for prokaryote initiation and termination of transcription
|
|
|
What is the product of eukaryotic RNA polymerase I, II and III
|
RNA pol I: 28s, 18s, 5.8s rRNA; RNA pol II: hnRNA/mRNA, snRNA; RNA pol III: tRNA, 5s rRNA
|
|
|
Steps in prokaryotic production of RNA
|
1. Sigma factor + RNA polymerase binds promoter TATA; 2. Transcription 5'-3' begins and sigma factor released; 3. Rho-independent or dependant termination.
|
|
|
Steps in eukaryotic production of RNA
|
1. RNA polymerase II binds promoter region with help of transcription factors. 2. RNA pol II transcribes introns and exons; 3. Ends transcription upon reaching termination sequence.
|
|
|
Steps of eukaryotic mRNA processing
|
1. Addition of 7-methylguanosine cap to the 5' end (ribosome binding site); 2. Poly-A tail attached to 3' end (aids transport to cytoplasm); 3. Splicing of introns by spliceosomes (snRNA)
|
|
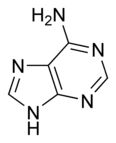
What is this structure? and what are the carbon donors?
|
Purine: Adenine. the middle C-C-N --> glycine; the tip of the molecule --> formate and glutamine; the carbon opposing a nitrogen in fenol ring --> CO2; the opposing N --> glutamine; remaining N --> aspartate; remaining C --> formate
|
|
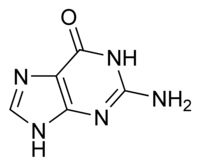
What is this structure?
|
Purine: Guanine
|
|
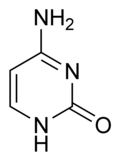
What is this structure and what are the carbon donators for synthesis?
|
Pyrimidine: Cytosine. Aspartate donates N-C-C-C; Carbamoyl phosphate (glutamine + CO2) donates N-C
|
|
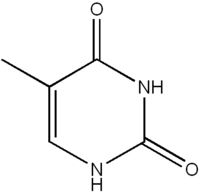
What is this structure?
|
Pyrimidine: Thymine
|
|
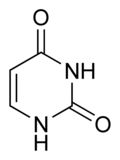
What is this structure?
|
Pyrimidine: Uracil
|
|
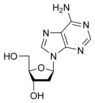
What is this structure?
|
Nucleoside
|
|
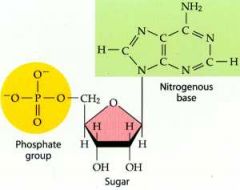
What is this structure?
|
Nucleotide
|
|
|
What is a silent mutation?
|
New codon codes same amino acid
|
|
|
What is a missense mutation?
|
New codon codes different amino acid
|
|
|
What is a nonsense mutation?
|
New codon codes a stop codon
|
|
|
What is a frameshift mutation?
|
Deletion or addition of a base
|
|
|
What is a large segment deletion? Mention two examples
|
Unequeal crossover in meiosis. Ex. Alpha thalasemia: deletion of alpha globin gene from chromosome 16; Cri-du-chat: 5q deletion
|
|
|
What is a triplet repeat expansion? Mention an example
|
Protein is longer than normal and unstable. Ex. Huntington CAG repeat codes multiple glutamines; also Fragile-X
|
|
|
Describe amino acid activation
|
1. AA + ATP + tRNA + aminoacyl-tRNA synthetase --> aminoacyl-tRNA + AMP + ppi. AAtRNA synthetase recognizes anticodon sequence of tRNA
|
|
|
Describe translation by activated aminoacyl-tRNA
|
Anticodon sequence of tRNA binds to codon on mRNA (antiparallel and complimentary)
|
|
|
What is a peptide bond?
|
Bond between carboxyl group of one AA to amino group of another AA
|
|
|
Protein synthesis: describe initiation process
|
1. Small subunit of ribosome binds Shine-Dalgarno or 5' cap of mRNA and slides down to first AUG codon; 2. tRNA binds start codon (methionine); 3. Large subunit binds small subunit
|
|
|
Protein synthesis: describe elongation process
|
1. Charged tRNA binds A site and mRNA codon; 2. Peptide bond by peptydyl transferase (uses 2 high energy bonds from aminoacyl-tRNAs); 3. tRNA is removed from P site/growing peptide; 4. ribosome moves 5'-3' exactly one codon. Translocation requires EF-2 (inactivated through ADP-rybosylation by Pseudomona and diptheria toxins)
|
|
|
Protein synthesis: describe termination process
|
1. Stop codon of mRNA (UGA, UAG, UAA) moves to A site; 2. peptydyl transferase releases peptide chain from tRNA in P site; 3. ribosome dissociates
|
|
|
What are the 4 structures formed during protein folding?
|
1. Primary structure: sequence of AA; 2. Secondary: stable a-helix and b-pleaded sheets; 3. Tertiary: interaction between secondary structures and final protein structure; 4. Cuaternary: multiple subunit interaction
|
|
|
What is the function of ubiquitin?
|
Covalently binds to misfolded proteins to signal their destruction by proteasome
|
|
|
Signal sequence required by proteins destined to be secreted, placed on cell membrane or directed to lysosome
|
N-terminal hydrophobic signal sequence
|
|
|
What is O-linked glycosylation?
|
Proteins acquire oligosacchride side chains attached to serine or threonine residues
|
|
|
What is N-linked glycosylation?
|
Proteins acquire oligosacchride side chains attached to asparagine residues
|
|
|
What does phosphorylation of manose residues do? I-cell disease?
|
Manose residues on oligosacchride chain are phosphorylated in golgi aparatus to direct the enzyme to lysosome. In I-cell disease, lysosomal enzymes are secreted into extracellular space due to inappropriate phosphorylation of manose residues
|
|
|
Mention 4 post-translational covalent modifications
|
1. Glycosylation: addition of oligosacchrides; 2. phosphorylation: addition of phosphate by kinases; 3. proteolysis: cleavage and activation of peptide bonds (proinsulin, trypsinogen, prothrombin); 4. γ-carboxylation: produces Ca+ binding sites (vitamin K dependant)
|
|
|
Describe postranslational modifications of collagen
|
1. N-terminal hydrophobic signals are added to prepro-alpha chains; 2. hydrophobic signal is removed in RER; 3. Hydroxylation of prolines and lysines (requires vit C); 4. Glycosylation of hydroxylysines; 5. Triple helical structures self-assemble (procollagen); 6. Secretion of procollagen; 7. Cleavage of propeptides; 8. Cross-linking of collagen by lysil oxidase (requires O2 & Cu+); 9. Aggregation of fibrils into collagen fibers
|
|
|
Biochemical defect in Scurvy
|
Deficient hydroxylation of pre-alpha collagen chains secondary to vitamin C defficiency. Vitamin C is required by lysyl and prolyl hydroxylases
|
|
|
Biochemical defect in Menkes disease
|
Deficient cross-linking of collagen secondary to functional copper deficiency: depigmented hair, arterial tortuosity, osteoporosis, anemia
|
|
|
Biochemical defect in Osteogenesis imperfecta
|
Mutations in collagen genes: skeletal deformities, fractures, blue sclrera
|
|
|
Biochemical defect in Ehlers-Danlos
|
Mutations in collagen genes and lysine hydroxylase gene: hyperextensible fragile skin, hypermobile joints, dislocations
|
|
|
What is an operon?
|
Group of prokaryote genes coding for a group of proteins required for a metabolic function, along with regulatory regions that control gene expression
|
|
|
The DNA sequence to which activator proteins bind in eukaryotes
|
Response elements
|
|
|
Mention 3 upstream promoter elements
|
1. CCAAT box (-75) that binds transcription factor NF-1; 2. TATA box; 3. GC-rich sequence that binds SP-1
|
|
|
The DNA regulatory base sequences in the vicinity of genes
|
cis regulators
|
|
|
What are trans regulators?
|
The transcription factors and the genes that code for them
|
|
|
Mention 4 characteristics of enchancers
|
1. Up to 1,000 bases away from gene; 2. Located upstream, downstream or within introns; 3. The orientation is unimportatnt; 4. Tissue specificity if transcription factors are not present in tissue
|
|
|
What does the activation domain of transcription factors allows them to do?
|
Interact with RNA polymerase II to stabilize the formation of the initiation complex
|
|
|
Mention 3 general transcription factors
|
SP-1, NF-1, TFIID (TATA factor)
|
|
|
Mention 3 specific transcription factors
|
steroid receptors, CREB protein PPARs
|
|
|
Describe the control of gluconeogenesis by response elements
|
1. Cortisol + zinc finger receptor bind GRE of PEPCK gene; 2. Glucagon increases cAMP --> protein kinase A --> phosphorylation of CREB --> CREB binds CRE --> upregulation of PEPCK. PEPCK converts OAA into PEP
|
|
|
What does acetylation of histones do?
|
Increases gene expression
|
|
|
What does methylation of DNA do?
|
Silences genes
|
|
|
What is genetic imprinting?
|
Results in mono allelic expression (Prader-Willi, Angelman)
|
|
|
What does glucose do to lactose operon in prokaryotes?
|
Increases intracellular cAMP, which inactivates CAP (activator protein), which decreases transcription of operon.
|
|
|
What does lactose do to the lactose operon in prokaryotes?
|
Inactivates active repressor and allows b-galactosidase gene transcription
|
|
|
What are palindromes?
|
Sequences of 4-8 bases that are inverted repeats and are recognized by restriction endonucleases. Example: GAATTC
|
|
|
What should a vector for recombinant DNA contain to be expressed?
|
Plasmid with restriction site, resistance to antibiotics gene, promoter and Shrine-Dalgarno sequence
|
|
|
Genomic DNA Vs. cDNA cloning
|
Genomic DNA: cleaved by restriction endonucleases, total DNA is cloned, introns; cDNA: reverse transcription of mRNA, genes expressed cloned, no introns
|
|
|
Type of material analyzed by Southern blot? Probe?
|
DNA. 32P-DNA probe. Used to determine restriction fragments of genes
|
|
|
Type of material analyzed by Northern blot? Probe?
|
RNA. 32P-DNA probe. To measure sizes and amounts of mRNA
|
|
|
Type of material analyzed by Western blot? Probe?
|
Proteins. 125I or antibody. To measure amount of antigen or antibody
|
|
|
AA precursors of catecolamines
|
Tyrosine, phenylalanine
|
|
|
AA precursor of serotonin
|
Tryptophan
|
|
|
What are the branched-chain AA
|
Valine, leucine, isoleucine
|
|
|
What are the acidic, negatively charrged AA
|
Aspartate, glutamate
|
|
|
What are the positively charged basic AA
|
Lysine, arginine, histidine
|
|
|
AA used as site for O-linked glycosylation
|
Serine, threonine
|
|
|
AA used as site for N-linked glycosylation
|
Asparagine
|
|
|
Sulfur-containing AA
|
Cysteine, methionine
|
|
|
Essential AA
|
Arginine, histidine, leucine, isoleucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine
|
|
|
What is Vmax?
|
The maximum rate possible to achieve with a given amount of enzyme
|
|
|
What is Km?
|
The substrate concentration required to produce half of Vmax
|
|
|
What does a high Km mean? Low Km?
|
High Km = low affinity; low Km = high affinity
|
|
|
What effect do competitive inhibitors have on Km?
|
Increase Km
|
|
|
What effect do noncompetitive inhibitors have on Vmax?
|
Decrease Vmax
|
|
|
What effect does increasing the enzyme concentration have on Vmax?
|
Increase Vmax
|
|
|
Mechanism of action: Gs receptor
|
1. Activates adenyl cyclase; 2. Increased cAMP; 3. Phosphorylation of protein kinase A and CREB
|
|
|
Mechanism of action: Gi receptor
|
1. Inhibits adenyl cyclase; 2. Decreases cAMP
|
|
|
Mechanism of action: Gq receptor
|
1. Activates phospholipase C; 2. Releases IP3 and DAG from membrane; 3. IP3 releases intracellular Ca+; DAG activates protein kinase C
|
|
|
Mechanism of action: tyrosine kinase receptor
|
1. Receptor binding by insulin, EGF, PDGF activates intrinsic tyrosine kinase on intracellular domain; 2. Insulin receptor substrate binds tyrosine kinase; 3. Activates SH2 domain proteins: PI-3 kinase (translocation of GLUT-4), p21RAS
|
|
|
Mechanism of action: 1,25 DHCC
|
Binds its zinc finger intracellular receptor in intestinal cells, which binds response elements in enhancer regions to induce synthesis of calcium binding proteins
|
|
|
Synthesis of vitamin D
|
1. UV light activates D3; 2. Liver 25 hydroxylase --> 25-HCC; 3. Kidney 1α-hydroxylase induced by PTH --> 1,25 DHCC
|
|
|
Biochemistry of vision
|
1. When rhodopsin Gt receptor is not stimulated by light, there are high levels of GMP in the rod cell. GMP activates Na-gated channels which partially depolarize the rod, releasing inhibitor glutamate on bipolar cells of optic nerve. 2. Upon light stimulation, rhodopsin Gt receptor decreases GMP, hyperpolarizes the cell and stops glutamate inhibition of bipolar cells
|
|
|
Enzymatic action of γ-glutamyl carboxylase
|
Introduces Ca+ binding sites by vitamin K-dependant carboxylation of glutamic acid
|
|
|
Mention the vitamin K-dependant factors
|
Factors II (prothrombin), VII, IX, X
|
|
|
Transport kinetics of GLUT-1
|
Low Km. At normal glucose concentrations GLUT-1 is at Vmax. Mediates glucose uptake in most tissues
|
|
|
Transport kinetics of GLUT-3
|
Low Km. At normal glucose concentrations GLUT-1 is at Vmax. Mediates glucose uptake in most tissues
|
|
|
Transport kinetics of GLUT-2
|
High Km. When glucose concentration drops below Km, the remainder glucose leaves the liver and enters the peripheral circulation. Located in hepatocytes and beta-cell.
|
|
|
Transport kinetics of GLUT-4
|
Insulin upregulatess expression of GLUT-4, increasing Vmax. Located in adipose tissue and muscle
|
|
|
Compare glucokinase Vs. Hexokinase
|
Both trap glucose into the cell by phosphorylation. Hexokinase is expressed by most tissues, has low Km, glucose-6-phosphate inhibits it (raises Km). Glucokinase is found in the liver and pancreas β-cell, high Km, induced by insulin in hepatocytes (increase Vmax)
|
|
|
Under which influence do phosphofructokinase inhibit the rate of glycolysis
|
Under the influence of glucagon (PFK-2) (increase cAMP/protein kinase A/phosphorylation) and ATP/citrate (PFK-1). Phosphorylated states noncompetitively inhibit PFK, lowering Vmax, lowering rate of glycolysis
|
|
|
PFKs are stimulated under the influence of what substances
|
Insulin-mediated dephosphorylation (PFK-2), AMP (PFK-2), fructose 2,6BP (PFK-1). Increase Vmax and rate of glycolysis
|
|
|
Rate-limiting enzyme in glycolysis
|
PFK-1, converts fructose-6P into fructose-1,6BP
|
|
|
What effect does increasing β-cell glucokinase Km have on glucose metabolism? Decrease Km?
|
An increased glucokinase Km (gene mutations), decreases production of insulin (MODY) and hyperglycemia because low glucose concentration in the cell (GLUT-2 has high Km) are not phosphorylated, thus cell doesn’t sense glucose and doesn’t produce insulin. A decreased glucokinase Km produces insulinemia and hypoglycemia.
|
|
|
Glyceraldehyde 3-P dehydrogenase
|
Oxidation of Glyceraldehyde-3P and reduction of NAD. Produces 1,3-BPG and NADH
|
|
|
3-PG kinase
|
Phosphorylates ADP using energy from 1,3-BPG. Produces ATP and 3-PG.
|
|
|
Pyruvate kinase
|
Substrate-level phosphorylation of ADP using energy from PEP. Produces pyruvate and ATP. Activated by fructose-1,6BP.
|
|
|
Lactate dehydrogenase
|
Oxidation of NADH producing lactate and NAD from pyruvate
|
|
|
Mention 3 irreversible reactions in glycolysis
|
1. Glucose + glucokinase/hexokinase --> glucose-6P; 2. Fructose-6P + PFK-1 + ATP --> Fructose-1,6BP; 3. PEP + pyruvate kinase --> pyruvate + ATP
|
|
|
Mention the 2 substrate-level phophorylations in glycolysis
|
1. 1,3BPG + phosphoglycerate kinase --> 3PG + ATP; 2. PEP + pyruvate kinase --> pyruvate + ATP
|
|
|
Reactions in the malate shuttle
|
OAA + NADH (cytoplasm) --> malate + NAD; malate enters mitochondria
|
|
|
Reactions in the glycerol-3P shuttle
|
DHAP + NADH --> glycerol-3P + glycerol-P dehydrogenase + FAD --> FADH2 (inner membrane/ETC/CoQ)
|
|
|
Substrates used for substrate-level phosphorylations in glycolysis
|
1,3-BPG, PEP
|
|
|
Oxygen dissociation curve behavior in high altitude
|
Low PO2 produces hyperventilation and respiratory alkalosis which shifts curve to the left (lowers p50). Rate of glycolysis increases producing more 2,3-BPG (12-24 hours) shifting the curve to normal p50
|
|
|
Effect of pyruvate kinase deficiency on RBC
|
2,3-BPG and p50 increase, Hb may not be fully saturated in the lungs. Decrease in ATP stimulates PFK-1 in RBC and rate of glycolysis increaes. RBC looses biconcave shape and is hemolised by spleen. Na/K ATPase activity decreases causing osmotic fragility and lysis.
|
|
|
Metabolism of galactose
|
Lactose -- glucose + galactose + galactokinase --> galactose-1P + gal-1P-uridyl transferase --> glucose-1P
|
|
|
Pathophysiology of cataracts in galactosemia
|
Galactose is converted to galactiol by aldose reductase in the lens. In diabetes glucose is also converted to sorbitol by the same enzyme.
|
|
|
Pathophysiology of hyperbilirubinemia in galactosemia
|
If galactokinase is present, galactose-1P is trapped in hepatocyte causing cirrohsis
|
|
|
Deficiency of aldolase B
|
Fructose-1P (produced by fructokinase) gets trapped in hepatocyte and proximal tubule producing jaundice and renal failure
|
|
|
Pyruvate dehydrogenase
|
Converts pyruvate to acetyl-CoA in the mitochondria for citric acid cycle and fatty acid synthesis. Requires NAD, FAD, CoA and thiamine. Negative feedback by acetyl-CoA.
|
|
|
Thiamine deficiency
|
Vitamin B1. Impairs citric acid cycle/glucose oxidation in highly aerobic tissue first (brain and heart) because of decreased pyruvate dehydrogenase activity and alpha-ketoglutarate dehydrogenase. Ataxia, nystagmus, memory loss, confabulation psychosis, cerebral hemorhage
|
|
|
Rate-limiting enzyme of oxidative phosphorylation
|
Isocitrate dehydrogenase. Inhibited by NADH and ATP, activated by ADP.
|
|
|
Synthesis of α-ketoglutarate
|
Isocitrate + NAD + isocitrate DH --> α-ketoglutarate + NADH + CO2
|
|
|
Enzymes that require thiamine, lipoic acid, NAD, FAD, CoA
|
Pyruvate DH, ɑ-ketoglutarate DH
|
|
|
Product of α-ketoglutarate DH
|
Succinyl CoA + NADH
|
|
|
Heme synthesis requires the product of this reaction
|
a-ketoglutarate + NAD + a-ketoglutarate DH --> succinyl CoA + NADH + CO2
|
|
|
Substrate-level phosphorylation reaction of oxidative phosphorylation
|
Succinyl CoA + GDP + succinyl CoA synthetase --> succinate + GTP
|
|
|
FADH2 is produced in this reaction of oxidative phosphorylation
|
Succinate + FAD + succinate DH (complex II) --> fumarate + FADH2
|
|
|
Substrate from oxidative phosphorylation used for fatty acid synthesis
|
Citrate (citrate shuttle)
|
|
|
Malate shuttle reaction in oxidative phosphorylation
|
Malate + NAD + malate DH --> OAA + NADH
|
|
|
Order of oxidative phosphorylation intermediate synthesis
|
Citrate, isocitrate, a-ketoglutarate, succinyl CoA, succinate, fumarate, malate, OAA
|
|
|
High-energy intermediates that produce NADH in oxidative phosphorylation
|
Isocitrate, a-ketoglutarate, malate
|
|
|
Complex I of ETC. Activators and inhibitors.
|
NADH dehydrogenase. Activated by NADH, inhibited by barbiturates and rotenone.
|
|
|
Complex II of ETC
|
Succinate dehydrogenase
|
|
|
Electron donors to CoQ
|
Succinate DH (FADH2), glycerol-P shuttle (FADH2), fattyacyl CoA DH (FADH2), NADH dehydrogenase (electron)
|
|
|
Complex III of ETC
|
Cytochrome b/c1
|
|
|
Complex III passes electrons to this protein
|
Cytochrome C
|
|
|
Complex IV of ETC. inhibitors, cofactors.
|
Cytochrome oxidase (cyt a/a3). Inhibited by cyanide, CO. Requires Cu+.
|
|
|
Mechanism of action of ETC uncouplers
|
Increase permeability to H+ decreasing the proton gradient of inner mitochodrial membrane. Decreases ATP synthesis, increases O2 consumption and oxidation of NADH, releases heat.
|
|
|
Uncouplers
|
2,4-dinitrophenol, aspirin and salicylates, UCP (thermogenin) of brown adipose tissue.
|
|
|
Mechanism of ATP synthesis in ETC
|
Energy from a proton passing through Fo inner membrane channel is used by F1 component (ATP synthetase) to phosphorylate ADP. (Proton gradient has potential kinetic energy)
|
|
|
ATP yielded by NADH and FADH2
|
NADH yields 3 ATP. FADH2 yields 2 ATP.
|
|
|
ATP per glucose yielded by anaerobic glycolysis
|
2 ATP/glucose
|
|
|
ATP per glucose yielded by aerobic glycolysis via malate shuttle
|
Yields 8 ATP/glucose
|
|
|
ATP per glucose yielded by aerobic glycolysis via glycerol-P shuttle
|
Yields 6 ATP/glucose
|
|
|
Effect of tissue hypoxia on ETC and cell
|
Hypoxia decreases the rate of ETC and ATP production, leading to anaerobic glycolysis, lactic acidosis and cell membrane damage due to decreased ATPase activity
|
|
|
Outcome of ETC inhibition
|
1. Decreased oxygen consumption; 2. Increased intracellular NADH and FADH2; 3. Decreased ATP
|
|
|
Mechanism of action of cyanide inhibition of ETC
|
Cyanide irreversibly binds to cytochrome a/a3
|
|
|
What are sources of cyanide?
|
Burning polyurethane. Byproduct of nitroprusside (thiosulfate can be used to destroy cyanide).
|
|
|
Antidote for cyanide poisoning
|
Nitrites convert hemoglobin to methhemoglobin which binds cyanide in the blood before it reaches tissues
|
|
|
Mechanism of CO poisoning
|
Binds cytochrome oxidase and displaces oxygen from hemoglobin
|
|
|
Important reactions in oxidative stress
|
O2 + NADPH oxidase --> superoxide radical
superoxide radical + superoxide dismutase --> H2O2 H2O2 + catalse --> H2O + O2 H2O2 + Cl- + myeloperoxidase --> hypochlorite |
|
|
How does cyanide poisoning inhibit glycolysis?
|
Complex IV is inhibited, NADH acumulates and inhibits isocitrate DH, citrate acumulates and inhibits PFK-1
|
|
|
Name of the core glycogen protein
|
Glycogenin
|
|
|
Metabolic regulation of glycogen synthetase
|
Activated by glucose and insulin (in liver) and insulin (in muscle). Inhibited by glucagon and epinephrine (in liver) and epinephrine (muscle)
|
|
|
Activation of glucose
|
Glucose 1-P --> UDP-glucose. Irreversible reaction
|
|
|
Synthesis of glycogen
|
Linear chain of glucose residues with alpha 1,4 bonds created by glycogen syhtetase. Branches by branching enzyme hydrolization of alpha 1,4 bonds and attachment with alpha 1,6 bond
|
|
|
Metabolic regulation of glycogen phosphorylase
|
Activated by epinephrine and glucagon (in liver). Epinephrine, AMP and Ca+ (in muscle). Inhibited by insulin (liver), insulin and ATP (in muscle)
|
|
|
Reactions of glycogenolysis
|
Glycogen phosphorylase irreversibly hydrolizes alpha 1,4 bonds releasing glucose 1-P. Debranching enzyme hydrolizes alpha 1,4 bonds adjacent to branch and moves oligoglucose to main chain then hydrolizes the alpha 1,6 bond releasing a free glucose. Glucose-6-phophatase converts glucose-6P into glucose.
|
|
|
Glucose 6 phosphatase
|
Converts glucose 1-P to glucose 6-P
|
|
|
In which condition is glycogen synthesis active in spite of high levels of glucagon and epinephrine?
|
Von Gierke disease. Profound hypoglycemia, so glucagon is high but glucose-6P stimulates glycogenesis.
|
|
|
Von Gierke disease
|
Deficiency of glucose 6 phosphatase. Severe hypoglycemia, lactic acidosis, hepatomegaly, hyperuricemia
|
|
|
McArdle's disease
|
Muscle glycogen phosphorylase deficiency. Exercise intolerance, muscle cramping, myoglobinuria.
|
|
|
Pompe disease
|
Lysosomal alpha 1,4 glucosidase deficiency. Cardiomegaly, glycogen inclusion bodies.
|
|
|
Substrates for gluconeogenesis
|
Lactate --> pyruvate
Alanine + ALT --> pyruvate Glycerol 3P from triacylglycerol |
|
|
Pyruvate carboxylase
|
Requires biotin. Activated by Acetyl-CoA. Produces OAA which leaves mitochondria via malate shuttle.
|
|
|
PEPCK
|
Induced by glucagon and cortisol. Converts OAA to PEP requiring ATP.
|
|
|
Fructose 1,6 biphosphatase
|
Hydrolizes phosphate from fructose 1,6BP. Activated by ATP, inhibited by AMP and fructose 2,6BP
|
|
|
Metabolic control of gluconeogenesis
|
Glucagon and cortisol induce PEPCK via GRE and CREB. Glucagon inhibits PFK-2, decreasing fructose 2,6BP a negative inhibitor of fructose 1,6 biphosphatse. Insulin activates PFK-2 increasing fructose 2,6BP which inhibits fructose 1,6, biphosphatase
|
|
|
Source of ATP for gluconeogenesis
|
From β-oxidation (gluconeogenesis depends on β-oxidation)
|
|
|
Regulation of pyruvate carboxylase and pyruvate DH by acetyl CoA
|
Acetyl CoA activates pyruvate carboxylase and deactivates PDH
|
|
|
Hypoglycemia induced by alcoholism
|
Alcohol metabolism produces excess NADH which interferes with gluconeogenesis by 1. favoring lactate formation from pyruvate; 2. Shifting the malate shuttle into the mitochondria; 3. Forming glycerol 3P from DHAP
|
|
|
Biochemistry of fatty liver in alcoholics
|
Fatty acids in the liver react with glycerol 3P to form triglycerides which get stored in the liver. The source of glycerol 3P is from impaired gluconeogenesis by NADH.
|
|
|
Why doesn't muscle produce blood glucose by gluconeogenesis?
|
It lacks glucose-6 phosphatase which is only in the liver
|
|
|
Reactions in alcohol metabolism
|
Alcohol + alcohol DH --> acetaldehyde + NADH + acetyldehyde DH --> acetate and NADH. Two NADH are released.
|
|
|
Rate-limiting enzyme of hexose-monophosphate shunt. Inhibitor and activators.
|
G6PDH. Induced by insulin, inhibited by NADPH, activated by NADP
|
|
|
Only thiamine-requiring enzyme in the red blood cell
|
Transketolase. Converts fructose 6P and glyceraldehyde into sugar intermediates for nucleotide synthesis in hexose-monophosphate shunt
|
|
|
Functions of NADPH
|
Biosynthesis. Maintenance of reduced gluthathione pool to protect against ROS. Bactericidal activity in PMN
|
|
|
Gluthathione reductase
|
Reduces gluthathione using NADPH
|
|
|
Gluthathione peroxidase
|
Oxidizes H2O2 using reduced gluthathione
|
|
|
Heinz bodies formation
|
H2O2 acumulates and denatures Hb because of lack of G6PDH and reduced gluthathione
|
|
|
Pathophysiology of G6PDH deficiency
|
Inadequate production of NADPH decreases the activity of gluthathione peroxidase which degrades H2O2 which denatures Hb (Heinz bodies) and peroxidate membrane lipids (hemolisis)
|
|
|
Chronic granulomatous disease
|
No production of superoxide radical in PMN to destroy catalase-positive organisms due to NADPH oxidase deficiency. Negative nitroblue tetrazolium test is diagnostic.
|
|
|
Important fatty acids
|
Linoleic acid: C18:2(9,12); Linolenic acid: C18:3(9,12,15); Arachidonic acid C20:4(5,8,11,14); Palmitic acid: C16:0
|
|
|
Trans fatty acids
|
Unnatural trans-double bonds are introduced by partial hydrogenation of vegetable oils to make them solid at room temperature (margarine). Associated with atherosclerosis
|
|
|
Cardio protective effects of Omega 3 fatty acids
|
Replaces some arachidonic acid in platelet membrane, decreasing production of platelet aggregator thromboxane A2
|
|
|
Fatty acid activation
|
CoA is attached by fatty acylCoA synthetase. Requires ATP.
|
|
|
Digestion of lipids
|
1. Bile emulsifies lipids; 2. pancreatic lipase, colipase and cholesterol esterase degrade lipids to 2-monoglyceride, fatty acids and cholesterol; 3. Absorbed and re-esterified to TG and cholesterol esters; 4. Packaged with Apo-B48 into chylomicrons
|
|
|
Regualtion of fatty acid synthesis by insulin
|
Increases production of acetyl CoA in the liver: glucokinase (induced), PFK-2/PFK-1 (PFK-2 dephosphorylated); Pyruvate DH (dephosphorylated). Fatty acid synthesis: acetyl CoA carboxylse (dephosphorylated), fatty acid synthetase (induced)
|
|
|
Citrate shuttle
|
Acetyl CoA + OAA + citrate synthase --> citrate --> cytoplasm --> cytrate + citrate lyase --> acetyl CoA + OAA. Acetyl CoA for fatty acid and cholesterol syhnthesis. OAA to malate for malate shuttle or pyruvate via malic enzyme
|
|
|
Malic enzyme
|
Converts malate into pyruvate releasing NADPH in cytoplasm for FA synthesis
|
|
|
Acetyl CoA carboxylase
|
Acetyl CoA + CO2 --> malonyl CoA in cytoplasm. Requires biotin. Activated by insulin and citrate. Rate-limiting in FA synthesis. The CO2 is not incorporated into FA because its removed by FA synthase.
|
|
|
Fatty acid synthase
|
8 malonyl CoA + NADPH --> palmitate + 8CO2. Requires panthothenic acid (B5 --> CoA). Induced by insulin in cytoplasm.
|
|
|
Cytochrome b5
|
Desaturation of fatty acids. Can't introduce double bonds past carbon 9 of FA.
|
|
|
How is acetyl coA activated for FA synthesis?
|
Its carboxylated to malonyl CoA by acetyl CoA carboxylase (activated by insulin)
|
|
|
Sources of glycerol 3P for triglyceride synthesis
|
Reduction of DHAP by glycerol 3P DH in adipose and liver; phosphorylation of glycerol by glycerol kinase in liver. Glycerol kinase works under insulin (VLDL metabolism) or glucagon (lypolysis glycerol) influence.
|
|
|
Glycerophospholipids
|
A glycerol with 2 FA and a water soluble group such as choline (phosphatidylcholine, lecithin) or inositol (phosphatidylinositol). Source for membrane-bound second messengers diacylglycerol, IP3, arachidonic acid. Needed for membrane synthesis
|
|
|
ApoB-48
|
Secreted by epithelial cells of intestine along with chylomicrons to allow their exit to lymph and tissues
|
|
|
Chylomicrons
|
Teansport dietary triglyceride and cholesterol from intestine to lymph to tissues. Have ApoB-48 plus ApoC-II and ApoE donated by HDL.
|
|
|
VLDL
|
Synthesized in liver to transport newly synthesized triglycerides to tissues. Have ApoB-100 and ApoC-II/ApoE donated by HDL
|
|
|
ApoC-II
|
Activates lipoprotein lipase. Donated to chylomicrons and VLDL by HDL
|
|
|
ApoE
|
Allows hepatocyte endocytosis of chylomicron and VLDL remnants
|
|
|
ApoA-1
|
Carried by HDL, activates LCAT and PCAT (blood enzymes) to hydrolize fatty streak cholesterol by attaching a fatty acid to it (esterification) so that it dissolves into HDL for reverse transport from tissues to hepatocyte
|
|
|
ApoB-100
|
Allows VLDL to exit from hepatocyte
|
|
|
Lipoprotein lipase
|
In the luminal surface of capillary endothelium. Releases fatty acids from triglycerides carried by chylomicrons and VLDL. Activated by ApoC-II and induced by insulin
|
|
|
IDL
|
VLDL remnant that is picked up by hepatocyte through ApoE receptor or acquire more cholesterol from HDL to become LDL
|
|
|
LDL
|
Delivers cholesterol to tissues for membrane and hormone synthesis. 80% picked up by hepatocytes to make bile acids. Clathrin endocytosis mediated by ApoB-100/LDL receptor
|
|
|
Cholesterol feedback loops
|
HMG-CoA reductase negative feedback. ACAT postive feedback. Represses expression of LDL receptor
|
|
|
HDL
|
Carry ApoA-1 which activates blood LCAT to esterize cholesterol and dissolve it into its core allowing reverse transport of cholesterol from the periphery to liver.
|
|
|
LCAT
|
Activated by ApoA-1from HDL. Binds a fatty acid to cholesterol to produce an ester that dissolves into the HDL for reverse transport to liver
|
|
|
CEPT
|
Transfers HDL cholesterol to IDL
|
|
|
SR-B1 receptors
|
Transfers HDL cholesterol into steroidgenic tissues ( ovaries, testes, adrenal, hepatocytes)
|
|
|
Pathophsysiology of atherosclerosis
|
1. Endothelial lesion produced by blood turbulence, elevated LDL, free radicals from cigarette smoke, homocystenemia, diabetes (glycosylation of LDL) and hypertension.; 2. Inflamed endothelium recruits monocytes and macrophages and platelet adhesion; 3. Production of ROS by macropahges oxidizes LDL; 4. Macrophages become cholesterol-laden (foam cells) after phagocytosis of LDL, producing fatty streaks; 5. Fatty streak enlarges with necrotic debris, lipids, epitheloid and smooth muscle cells producing an advanced plaque and ocluding blood vessel with subsequent ischemia; 6 The plaque can rupture with subsequent thrombosis
|
|
|
Vitamin E
|
Only lipid-soluble antioxidant, protects LDL from oxidation and peroxidation of membranes by ROS
|
|
|
MCC of elevated triglyceride and chylomicrons
|
Type IV hyperlipidemia in diabetics. Low insulin levels fail to induce lipoprotein lipase
|
|
|
Familial lipoprotein lipase or ApoC-II deficiency
|
Type I hypertriglyceridemia. Red-orange xanthomas, fatty liver, acute pancreatitis, abdominal pain
|
|
|
LDL receptor deficiency
|
Type Iia familial hypercholesterolemia. Autosomal dominant. Atherosclerosis and CAD, xanthomas of achilles tendon, tuberous xanthomas, xanthelasmas, corneal arcus.
|
|
|
MCC of hypolipidemia
|
Abetalipoproteinemia (deficiency of ApoB-48 and ApoB-100). No serum chylomicrons, triglycerides or cholesterol. ADEK vitamin deficiency, steatorrhea, ataxia, pigmentary degeneration of retina, acanthocytes, loss of night vision
|
|
|
DeNovo cholesterol synthesis
|
2 Aacetyl CoA provided by citrate shuttle + NADPH --> acetoacetyl CoA + HMG-CoA synthase --> HMG-CoA + HGM-CoA reductase --> mevalonate -->--> farnesyl ppi -->--> cholesterol.
|
|
|
Farnesyl ppi
|
Cholesterol synthesis intermediate important for synthesis of CoQ, dolichol ppi for N-linked glycosylation and prenylation
|
|
|
Rate-limiting enzyme in cholesterol synthesis
|
HMG-CoA reductase converts HMG-CoA to to mevalonate. Insulin activates it by dephosphorylation, gene expression repressed by cholesterol, competitively inhibted by statins
|
|
|
MOA of cholestyramine
|
Increases elimination of bile salts, lowering cholesterol levels in hepatocyte which increases LDL receptor expression and clearing of LDL from blood
|
|
|
MOA of statins and important side effects
|
Increase HMG-CoA reductase Km. Decreases denovo cholesterol synthesis in hepatocyte which increases LDL receptor expression and clears LDL from blood. Side effects: decreased farnesyl ppi decreases synthesis of CoQ, decreasing ATP production. Myopahty, rhabdomyolisis, mioglobimuria
|
|
|
Hormone-sensitive lipase
|
Hydrolizes triglycerides to glycerol and fatty acids in adipose tissue. Activated by decreased insulin, increased epinephrine and cortisol. Inhibited by niacin.
|
|
|
Fatty acyl CoA synthetase
|
On the outer mitochondrial membrane, activates fatty acids by attaching CoA
|
|
|
Carnitine shuttle
|
CAT-1 on outer mitochindrial membrane tranfers fattyacyl group of activated fatty acid to carnitine. Carnitine transporter shuttle fattyacyl-carnitine into inner mitochondrial matrix. CAT-2 on inner membrane transfers fattyacyl to CoA. CAT-1 Is inhibited by malonylCoA from fatty acid synthesis
|
|
|
Why don’t fatty acids enter mitochondria after FA synthesis?
|
Under influence of insulin, increased acetyl CoA carboxylase produces malonyl CoA which inhibits CAT-1
|
|
|
fatty acyl CoA dehydrogenase
|
Enzyme of B-oxidation in mitochondrial matrix. Oxidizes activated fatty acids to acetyl CoA producing FADH2 (for CoQ) and NADH (for NADH DH). Also called LCAD and MCAD. Inhibited by Ackee fruit.
|
|
|
Myopathic CAT deficiency
|
No FA for B-oxidation. Muscle aches and weakness, rhabdomyolisis, myoglobinuria, elvated muscle TG
|
|
|
rhabdomyiolysis, mioglobinuria, muscle weakness
|
Can be produced by either farnesyl ppi deficiency from cholesterol intermidiate which is needed for CoQ synthesis and ETC, or by CAT deficiency which inhibits carnitine shuttle and B-oxidation
|
|
|
Cause of non-ketotic hypoglycemia
|
MCAD deficiency. No B-oxidation, decereased ATP for gluconeogenesis, hypoglycemia, decreased acetyl CoA lowers pyruvate carboxylase activity and limits ketogenesis. Profound fasting non ketotic hypoglycemia, dicarboxilic acidemia, C8-C10 acyl carnitines in the blood
|
|
|
True or false: fatty acids cannot be converted to glucose
|
True with exception: Odd carbon FA produce propionyl CoA which is converted to succinyl CoA and leave as malate to cytoplasm for gluconeogenesis
|
|
|
Enzyme requires B12
|
Methylmalonyl CoA mutase converts methylmalonyl CoA into succinyl CoA in propionic acid pathway. Deficiency of B12 produces megaloblastic anemia and methylmalonic aciduria
|
|
|
Propionyl CoA carboxylase
|
Converts propionyl CoA from odd-carbon β-oxidation into methylmalonyl CoA
|
|
|
What are the ketone bodies and what tissues metabolize them?
|
Acetoacetate and B-hydroxybutirate metabolized by muscle, cardiac muscle and renal cortex. Brain metabolizes them after 1 week of fasting
|
|
|
Important intermediate of ketogenesis
|
HMG-CoA produced from acetoacetyl CoA by HMG-CoA synthase
|
|
|
HMG-CoA lyase
|
Converts HMG-CoA into acetoacetate in ketogenesis
|
|
|
Why cant the liver metabolize ketones?
|
It lacks succinyl CoA-acetoacetyl CoA transferase (thiophorase)
|
|
|
Why and when does brain switch to ketogenolysis?
|
After one week of fasting acetyl CoA production from ketogenolysis inhibits pyruvate DH
|
|
|
Milestones in brain energy metabolism
|
12 hours: glucose from glycogen; 1 week: glucose from gluconeogenesis; beyond 1 week: ketone bodies
|
|
|
Who develops ketoacidosis?
|
Type 1 and Type 2 diabetics and alcoholics
|
|
|
How does an infection triger ketoacidosis in diabetics?
|
Increased cortisol and epinephrine activate hormone-sensitive lipase and increases B-oxidation and ketogenesis
|
|
|
Pathophysiology of alcoholic ketoacidosis
|
Chronic hypoglycemia favors ketogenesis in liver but muscle would rather use acetate from alcohol metabolism which increases ketoacids in blood
|
|
|
Urinary nitroprusside test
|
Detects acetoacetate in urine which could underestimate B-hydroxybutirate in ketoacidosis
|
|
|
Signs and symptoms of ketoacidosis
|
Polyuria, polydypsia (hyperglycemia and osmotic diuresis), coma, depletion of K+ (masked by hyperkalemia), decreased HCO3 fruity odor
|
|
|
Cherry-red spots in macula, blindness, psychomotor retardation, death before 2 years
|
Tay SaXhH. Deficiency of Hexosaminidase A. Ganglioside acumulates
|
|
|
Hepatosplenomegaly, bone erosion, fractures, pancytopenia
|
Gaucher. Glucocerebroside deficiency. Glucocerebroside acumulates.
|
|
|
Cherry-red macula spots, hepatosplenomegaly, microcephaly, retardation
|
Nieman PickS: Sphingomyelinase deficiency. Sphingomyelin accumulates
|
|
|
Glutamine synthetase
|
Captures excess nitrogen by aminating glutamate to glutamine. Located in most tissues.
|
|
|
Glutaminase
|
Located in kidney and intestine. Deaminates glutamine releasing amonia. In kidney it's induced by chronic acidosis and amonia combines to form amonium and its excreted. In the intestine, amonia is sent to liver via portal blood where it enters the urea cycle
|
|
|
Amination of alpha-ketoglutarate
|
Produces glutamate and alpha-ketoacid by aminotransferase (requires B6 pyridoxine)
|
|
|
Amination of pyruvate
|
Produces alanine and alpha-ketoglutarate. Amino group donated by glutamate. Alanine aminotransferase requires B6 and its in muscle and liver
|
|
|
Amination of OAA
|
Produces aspartate for urea cycle in liver and alpha-ketoglutarate. Amino group donated by glutamate. Requires B6
|
|
|
Donation of amino group by glutamate produces what?
|
Donation to pyruvate produces alanine. Donation to OAA produces aspartate. Deamination by glutamate DH produces free amonia in liver for urea cycle
|
|
|
Donators of amonia to urea cycle
|
Intestinal glutamine is deaminated by glutaminase and goes to portal blood. Glutamate is deaminated by glutamine DH releasing free amonia
|
|
|
How does muscle nitrogen in alanine enter urea cycle?
|
Alanine donate to alpha-ketoglutarate to form glutamate which donates to OAA to form aspartate (in liver)
|
|
|
Glutamate DH
|
Deaminates glutamate to release free amonia for urea cycle and alpha-ketoglutarate for citric acid cycle
|
|
|
Non-glucogenic AA
|
Leucine, lysine (ketogenic)
|
|
|
Ketogenic and glucogenic AA
|
Phe, Tyr, Try, Ile, Thr
|
|
|
Mitochondrial urea cycle enzymes
|
Carbamoyl phosphate synthetase I, ornithine transcarbamoylase
|
|
|
Citrulline
|
Produced in mitochondria by ornithine transcarbamoylase using carbamoyl phosphate and ornithine. Leaves mitochondria and combines with aspartate in cytoplasm
|
|
|
Blood findings if there is a defect of the urea cycle
|
Hyperamonemia, elevated blood glutamine, decreased BUN
|
|
|
Carbamoyl phosphate
|
Substrate for ornithine transcarbamoylase (urea cycle) and aspartate transcarbamoylase in pyrimidine synthesis. If it acumulates it increases uracil and orotic acid in blood and urine (pyrimidine synthesis intermidiates)
|
|
|
This substance acumulates in ornithine transcarbamoylase deficiency
|
Carbamoyl phosphate. Produces hyperamonemia, decreased BUN, elevated glutamine, uracil and orotic acid
|
|
|
Sodium benzoate, phenylpyruvate
|
Treatment of urea cycle enzyme deficiencies. Provides alternative route for capturing and excreting excess nitrogen
|
|
|
Phenylketonuria
|
Phenylalanine hydroxylase deficiency. Mental retardation, microcephaly, pale skin, blonde hair, musty odor. Avoid aspartame, monitor pregnancy
|
|
|
Alkaptonuria
|
"Black Homo". Dark urine, ochronosis. Homogentisate oxidase deficiency.
|
|
|
Maple syrup urine
|
Branched-chain ketoacid dehydrogenase deficiency. Lethargy, alternating episodes of hypertonia and hypotonia, odor of maple syrup urine, ketosis, coma.
|
|
|
Neonatal ketoacidosis
|
Propionyl coa Carboxylase or methyl malonyl CoA mutase deficiency. Cant metabolize val, met, ile, thr through propyonyl acid cycle
|
|
|
Methylmalonic acidura
|
Methylmalonyl CoA mutase deficiency in propionic acid pathway
|
|
|
Reactions that require methyl groups
|
Epinephrine synthesis, N-methylguanosine cap on mRNA, synthesis of purines and thymidine
|
|
|
Methyl donors (one-carbon units)
|
SAM (S-adenosylmethionine); tetrahydrofolate
|
|
|
Activation of folate
|
Folate --> dihydrofolate + dihydrofolate reductase --> THF + 1-carbon unit --> active folate
|
|
|
Homocystinemia and homocystinuria
|
Produced by cysthathione synthase deficiency or homocysteine-methyl THF methyltransferase deficiency or folate/B12 deficiency. Cysteine becomes essential, decrease methionine intake. Cardiovascular disease, atherosclerosis, DVT, thromboembolism, ectopic lens
|
|
|
B12 deficiency
|
Megaloblastic anemia, neuropathy, homocystinemia, methylmalonic aciduria. Needed by methylTHF-homocysteine methyl transferase and malonyl CoA mutase. Pernicious anemia, D. latum, ileum resection, Crohn's
|
|
|
Folate deficiency
|
Megaloblastic anemia, homocystinemia. Found in alcoholism and pregnancy
|
|
|
What is the folate storage pool and what enzyme acts upon it?
|
Stored as reduced methylTHF. MethylTHF-homocysteine methyl transferase transfers methyl group to homocysteine to form methionine which produces one-carbon unit donor SAM
|
|
|
Vitamin B1
|
Thiamine. Deficiency --> Wernicke-Korsakoff, wet and dry Beri Beri. Required by dehydrogenases
|
|
|
Biotin
|
Required by carboxylases. MCC of deficiency is due to consumption of raw eggs (avidin)
|
|
|
Vitamin B2
|
Riboflavin. Precursor of FAD.
|
|
|
Vitamin B3
|
Niacin. Precursor to NAD and NADPH used by dehydrogenases.
|
|
|
vitamin B5
|
Panthotenic acid. Deficiency results in dermatitis, enteritis, alopecia.
|
|
|
Vitamin B6
|
Pyridoxine. Required for heme synthesis and transaminations
|
|
|
Pantothenic acid
|
Vitamin B5 precursor of CoA
|
|
|
B2 deficiency
|
Riboflavin. Deficiency results in angular stomatitis (inflamation of oral mucous lining), cheilosis (inflammation of lips), corneal vascularization.
|
|
|
Pellagra
|
Niacin (B3) deficiency results in diarrhea, dermatitis, dementia and glositis.
|
|
|
Wet (adult) beri beri
|
B1/thiamine deficiency. Heart failure (dilated cardiomyopathy) and edema (wet)
|
|
|
Dry (infantile) beri beri
|
B1/Thiamine deficiency. Muscle wasting, neuropathy, can also have cardiomegaly
|
|
|
Base excision repair
|
Glycosylase tags defective bases; endonuclease and lyase cleave out defective bases; DNA polymerase synthesizes new bases; ligase reseals the DNA.
|
|
|
Types of DNA damage
|
Thymine dimers (UV light); depurination (spontaneous or chemicals); breaks and oxidative damage (ionizing radiation); Cross-linkage, intercalation, alkylation (chemical/drugs)
|
|
|
Important enzymes in heme synthesis
|
ALA synthase (stimulated by alcohol, barbiturates, hypoxia) and ferrochelatase are in the mitochondria of immature RBCs. ALA dehydrase and ferrochelatase are denatured by lead.
|
|
|
What is responsible for higher O2 affinity of HbF?
|
Presence of serine instead of histidine in the 2,3BPG binding sites of the beta chains. Serine decreaes 2,3BPG binding to beta chains and increases affinity.
|
|
|
Metabolism of homocysteine
|
Can be converted to cysthathione by cysthathione synthetase and then cysteine (requires B6). Or it can be converted into methionine (requires B12 and 5-methylTHF)
|
|
|
HbC versus HbS
|
HbS is formed by replacement of glutamic acid by valine (neutral); HbC is formed by replacement of glutamic acid by lysine (positively charged). Therfore HbC moves less than HbS in electrophoresis.
|
|
|
Thymidilate synthetase
|
Makes dTMP which is the deNovo precursor of DNA nucleotides. Requires methylTHF
|
|
|
Cofactors required by dehydrogenases
|
FAD, NAD, thiamine, CoA, lipoic acid. Deficiencies of these results in lactic acidosis (piruvate DH) and maple syrup urine (branched-chain ketoacid DH)
|
|
|
Components of the LAC operon
|
Inducer (lactose), repressor (decreased by lactose), activator protein (activated by low cAMP/low glucose)
|
|
|
Which substance acumulates in lead poisoning?
|
delta-ALA (the substrate of delta-ALA dehydrase)
|
|
|
Bohr effect
|
High CO2 in tissues increases carbonic anhydrase synthesis of protons which bind to Hb and decrease affinity for oxygen (releasing it). In the lungs, high PO2 releases the protons from the ionizable histidine residues at N-terminals of alpha and beta chains
|
|
|
Enzymes that require biotin
|
Carboxylases. Biotin deficiency leads to increased pyruvate (pyruvate carboxylase) that is converted to lactate --> lactic acidosis
|
|
|
Gluconeogenic enzymes
|
Pyruvate carboxylase, Fructose 1-6 biphosphatase, PEPCK, glucose-6-phosphatase
|
|
|
Ketogenic enzymes
|
AcetoacetylCoA synthase, HMG-CoA synthase, HMG-CoA lyase
|
|
|
beta oxydation enzymes
|
CAT-1, CAT transporter, CAT-2, MCAD/LCAD (fattyacylCoA DH), fattyacylCoA synthase
|
|
|
Elastin
|
Responsible for lung recoil and elasticity of tissues. Composed of glycine, valine and alanine. Secreted as tropoelastin which crosslinks with fibrillin (needs lysyl oxidase). Elastase breaks down cross-links, alpha-1-antitrypsin inhibit elastases.
|
|
|
Receptors/second messengers used by cytochines
|
MAP/JAK/STATS (intrinsic tyrosine kinase activity)
|
|
|
Receptors/second messengers used by atrial natriuretic peptide
|
Guanylate cyclase pathway
|
|
|
Receptors/second messengers used by TSH, PTH, glucagon
|
Gs --> adenyl cyclase --> increase cAMP
|
|
|
Phosphodiesterase
|
Inactivates cAMP
|
|
|
Positively-charged Aas
|
Arginine, histidine, lysine (all essential)
|
|
|
Polar AAs
|
Ser, thr, cys, met, asn, gln
|
|
|
Aromatic AAs
|
Tyr, phe (essential), trp
|
|
|
N-acetylglutamate synthase
|
Makes N-acetyl glutamate which is the allosteric activator of carbamoyl phosphate synthase I
|
|
|
Cofactor for phenylalanine and tryptophan hydroxylases
|
Tetrahydropterin (BH4)
|
|
|
N-acetyl glutamate
|
allosteric activator of carbamoyl phosphate synthase I
|
|
|
Fructose metabolism
|
Bypases PFK-1 and therefore has the highest metabolic rate
|
|
|
Molecular defect in Marfan
|
Fibrillin defect. Fibrillin is necessary for elasticity of tissues and suspensory ligaments of the lens (ectopic lens)
|
|
|
Metabolism of branched-chain AAs
|
All require BCAA-DH. Val and Ile are metabolized via propionic acid path; Leu via acetyl CoA
|
|
|
Tyrosine
|
Needed for catecholamine and homogentisate synthesis
|
|
|
Vitamin A toxicity
|
Alopecia, dry skin, hepatomegaly, hyperlipidemia
|
|
|
AAs metabolized via propionic acid path
|
Val, Ile, Thr, Met
|
|
|
Zellweger syndrome
|
Peroxisome deficiency with lack of long-chain FA metabolism. Hypotonia, seizures, hepatomegaly, retardation.
|
|
|
Inducers and inhibitors of ALA dehydrase
|
Alcohol,barbiturates, hypoxia are activators; Heme inhibits it
|
|
|
Hartnup disease
|
Defective intestinal/kidney reabosorption of Try which is essential and required precursor of niacin. Pellagra-like symptoms and basic aminoaciduria
|
|
|
Telomerase
|
Reverse transcriptase adds TTAGGG telomers to chromosomes of pluripotent and dividing cells
|
|
|
Proof-reading activity
|
DNA polymerases have 3'-5' exonuclease proof-reading activity
|
|
|
5'-3' exonuclease activity
|
Only DNA polymerase I (removes primers)
|
|
|
Cysthathione synthase deficiency
|
Homocystinemia. DVT, atherosclerosis, ectopic lens. Cysteine becomes essential AA. Rx.: B6 and cysteine supplements
|
|
|
Functions of vitamin A
|
Retinal pigments, differentiation of epithelial tissues
|
|
|
Propionic acidemia
|
Propionyl CoA carboxylase deficiency. Poor feeding, vomiting, hypotonia, lethargy, dehydration, acidosis
|
|
|
Stop and start codons
|
AUG (start, methionine); UAA, UAG, UGA are stop codons
|
|
|
Pyruvate DH deficiency
|
hypoxia-induced lactic acidosis
|
|
|
Acute intermitent porphyria
|
Abdominal pain, neurologi manifestations, no photosensitivity, coluria, increased ALA and PB
|
|
|
Regulators of ALA dehydrase
|
Alcohol, barbiturates and hypoxia activate it; heme inhibits it
|
|
|
Enzyme deficiencies that result in photosensitivity
|
Uroporphyrinogen decarboxylase, coproporphyrinogen oxidase and ferrochelatase
|
|
|
Enzymes that require B1
|
Branched-chain DH, alpha-ketglutarate DH, pyruvate DH, transketolase
|
|
|
Causes of pellagra
|
Niacin is made from tryptophan and requires B6 - therefore pellagra can be caused by Hartnup, malignant carcinoid (increases try metabolism), isoniazid (decreased B6)
|
|
|
Vitamin B6 deficiency
|
Induced by isoniazid and contraceptives - peripheral neuropathy, seizures
|
|
|
Vitamin D excess
|
Results in hypercalcemia, stupor; Seen in sarcoidosis due to macrophage conversion of vit D into active form
|
|
|
Vitamin K deficiency
|
Seen in neonates because intestine have no flora to synthesize it; results in neonatal hemorrhage with normal bleeding time and increased PT and PTT
|
|
|
Vitamin K dependant factors
|
II, VII, IX, X and proteins C and S
|
|
|
Limitting factor in ethanol metabolism
|
NAD
|
|
|
Inhibitors of ethanol metabolism
|
Fomepizole (inhibits alcohol DH) and disulfram (inhibits acetaldehyde DH)
|
|
|
Kwashiorkor
|
Protein malnutrition --> edema, anemia, fatty liver
|
|
|
Marasmus
|
Total caloric malnutrition --> muscle wasting, loss of subcutaneous fat and edema
|
|
|
Bonds between purines and pyrimidines
|
G-C (3 H bonds) stronger than A-T bond (2 H bonds); increased G-C content --> increased melting temperature
|
|
|
Transition
|
Substituting pyrimidine for pyrimidine or purine for purine
|
|
|
Transversion
|
Substituting purine for pyrimidine or vice versa
|
|
|
Direction of replication and transcription
|
5'-3'. The incoming nucleotide bears the triphosphate in 5' that attaches to the 3' hydroxyl of nascent peptide
|
|
|
tRNA wobble
|
The third position of the codon may not be important for accurate base pairing
|
|
|
Energy requirements of translation
|
tRNA aminoacylation: ATP --> ADP; tRNA loading: GTP --> GDP; translocation: GTP --> GDP; total: 4 high-energy phosphate bonds
|
|
|
Enzyme regulation methods
|
Enzyme concentration alteration, covalent modification (phosphorylation), proteolytic activation, allosteric regulation, pH, temperature, transcriptional regulation
|
|
|
Permanent cells
|
Remain in G0, regenerate from stem cells; neurons, skeletal and cardiac muscle, RBCs
|
|
|
Stable quiescent cells
|
Enter G1 from G0 when stimulated; hepatocytes and lymphocytes
|
|
|
Labile cells
|
Never go to G0; bone marrow, gut epithelium, skin, hair follicles
|
|
|
Nissl bodies
|
RER of neurons
|
|
|
Dynein
|
ATPase that links peripheral 9 doublets to bend and slide cilium. Responsible for retrograde transport. Defective in Kartagener
|
|
|
Phosphatidylcholine (lecithin)
|
Component of RBC membranes, myelin, bile and surfactant. Used in sterification of cholesterol (LCAT)
|
|
|
Metabolic processes in the mitochondria
|
Beta oxydation, acetyl-CoA production, Krebs, ETC
|
|
|
Metabolic processes in the cytoplasm
|
Glycolysis, fatty acid synthesis, HMP shunt, protein synthesis (RER), steroid synthesis (SER)
|
|
|
Metabolic processes in mitochondria and cytoplasm
|
Heme synthesis, urea cycle, gluconeogenesis
|
|
|
Tissues that only carry anaerobic glycolysis
|
RBCs, leukocytes, kidney medulla, lens, testes, cornea
|
|
|
Gluconeogenic tissues
|
Liver, kidney, gut epithelium
|
|
|
Consequence of hyperammonemia
|
ornithinetranscarbamoyl synthase deficiency or liver disease --> hyperammonemia --> depletion of alpha-ketoglutarate --> inhibition of TCA cycle
|
|
|
Donators of NH2 and C to urea molecule
|
aspartate --> NH2; NH4+ --> NH2; CO2 --> C
|
|
|
Cystinuria
|
Defect of renal amino acid transporter for cysteine, lysine and arginine. Leads to cysteine stones. Rx.: acetazolamide
|
|
|
Adenosine deaminase deficiency
|
dATP accumulates which inhibits ribonucleotide reductase. Results in SCID due to decreased purine synthesis
|
|
|
Lesch Nyhan
|
Deficiency of HGPRT (hypoxanthine guanine phosphoribosyl pirophosphate transferase) which converts guanine to GMP and hypoxanthine to IMP. Hyperuricemia, retardation, self-mutilation, aggression, gout, choreoathetosis.
|
|
|
Pyrimidine synthesis
|
CO2 + glutamine + carbamoyl phosphate synthetase 2 --> carbamoyl phosphate + aspartate -->--> orotic acid + PRPP (from HMP shunt) --> UMP + ribonucleotide reductase --> dUMP + thymidylate synthase + THF --> dTMP + DHF
|
|
|
Dihydrofolate reductase
|
makes THF out of DHF for use of thymidylate synthase; inhibited by methotrexate (eukaryots) trimethorpim (prokaryotes)
|
|
|
Thymidylate synthase
|
makes dTMP out of dUMP in pyrimidine synthesis; inhibited by 5-fluoracil
|
|
|
Ribonucleotide reductase
|
Makes deoxyribonucletides from ribonucleotides. Inhibited by hydroxyurea
|
|
|
Carbamoyl phosphate synthase 2
|
Located in the cytoplasm. Makes carbamoyl phosphate from CO2 + glutamine in pyrimidine synthesis.
|
|
|
Pyrimidine synthesis major enzymes
|
Carbamoyl synthase 2, ribonucleotide reductase (inhibited by hydroxyurea), thymidylate synthase (inhibited by 5-FA), dihydrofolate reductase (inhibited by methotrexate and TMP)
|
|
|
Purine synthesis
|
Ribose-5-phosphate + PRPP synthase --> PRPP + PRPP amidotransferase --> 5-phosphoribosylamine + gly, asp, glu, THF -->-->--> IMP (contains hypoxanthine)
|
|
|
PRPP synthase
|
Makes PRPP from ribose-5-phosphate in purine synthesis.
|
|
|
PRPP amidotransferase
|
Makes 5-phosphoribosylamine from PRPP in purine synthesis. Inhibited by allopurinol and 6-mercaptupurine.
|
|
|
Why does Von Gierke and galactosemia produce hyperuricemia?
|
Excess consumption of Pi leads to accumulation of nucleosides that are degraded by xanthine oxidase
|
|
|
Enzymes of purine salvage pathway
|
Adenosine deaminase (deficient in SCID), xanthine oxidase, HPRT, HGPRT (deficient in Lesch Nyhan)
|
|
|
Heme synthesis
|
Glycine + succinyl CoA + ALA synthase --> δALA + ALA dehydrase --> porphobilinogen + uroporphyrinogen-I synthase --> uroporphyrinogen III -->-->--> protoporphyrin IX + Fe+ + ferrochelatase --> heme
|
|
|
Major enzymes of heme synthesis
|
ALA synthase (requires B6, negative feedback by heme); ALA dehydrase (inhibited by lead); uroporphyrinogen synthase (deficiency leads to acute intermittent porphyria); ferrochelatase (inhibited by lead)
|
|
|
Acute intermittent porphyria
|
Anxiety, confusion, paranoia, acute abdominal pain, no photosensitivity, coluria increased δ-ALA and porphobilinogen; due to uroporphyrinogen synthase deficiency; autosomal dominant, late onset, variable expression
|
|
|
↓protoporphyrin, ↓δ-ALA, ↑ferritin, ↑serum iron
|
Pyridoxine deficiency (isoniazid treatment)
|
|
|
↑protoporphyrin, δ-ALA normal, ↓ferritin, ↓serum iron
|
Iron deficiency anemia
|
|
|
↑protoporphyrin, ↑δ-ALA, ↑ferritin, ↑serum iron
|
Lead poisoning
|
|
|
phospholipids of the cell membrane
|
phosphatidylcholine, sphingomyelin, phosphatidylentholamine, phosphatidylserine
|
|
|
flippase
|
removes phosphatidylenotholamine and phosphatidylserine from the outer leaflet of the cell membrane using ATP
|
|
|
glycolipids of the cell membrane
|
cerebrosides, gangliosides (contains sialic acid), neutral glycolipids
|
|
|
NMDA receptor
|
require glutamate and glycine to allow Na and Ca influx and K efflux; irreversible antagonists: amantadine and memantine; reversible antagonists: ketamine and PCP
|
|
|
AMPA receptor
|
binds glutamate and allows Ca and Na influx and K efflux; responsible for excitotoxicity
|
|
|
L-type Ca channel
|
also called dyhydropiridine receptor; allows Ca influx and depolarization of cardiac and smooth muscle and interacts with ryanodine sarcoplasmic channels of smooth, cardiac and skeletal muscle
|
|
|
Fast Ca channel
|
also called ryanodine receptor; interacts with the L-type Ca channels to release Ca from the sarcoplasmic reticulum
|
|
|
T-type Ca channel
|
located in the SA and AV nodes (phase 4) and in the thalamus (ethosuximide)
|
|
|
nACh receptor
|
binds ACh and allow Na influx to depolarize muscle; succinylcholine (agonist, depolarizing) and tubocurarine/curonium's (antagonists, nondepolirizing)
|
|
|
GABAa receptor
|
two alpha subunits and 3 beta or gamma; 2 GABA molecules bind the two alpha subunits for influx of Cl
|
|
|
Gs receptor
|
has alpha, beta and gamma subunits; GDP is bound to the alpha subunit; binding of its substrate changes GDP to GTP and units dissociate and adenyl cyclase is activated to increase cAMP; alpha subunit deactivates GTP
|
|
|
protein kinase A
|
phosphorylates seine and threonine residues of intracellular enzymes to increase activity
|
|
|
zinc finger receptor
|
intracellular receptor made up of four cysteines and a zinc atom that has hormone-binding and DNA-binding regions; crosses nuclear membrane to directly influence transcription; TFIIA, Sp1, steroid hormone receptors
|
|
|
postranslational modifications
|
N-linked and O-linked glycosylation, phosphorylation of tyrosine, serine, threonine residues, sulfation, methylation and acetylation of lysine residues, gamma-carboxylation of glutamate, myristoylation of glycine, palmitoylation and fernasylation of cysteine; all happen in the Golgi complex
|
|
|
clathrin coated vesicles
|
for receptor-mediator endocytosis or packaging of proteins destined for secretion and lysosome
|
|
|
non-clathrin-coated vesicles
|
for proteins destined for the membrane
|
|
|
smooth endoplasmic reticulum
|
synthesis of membrane phospholipids, steroids, P450 and glucoronyl transferase enzymes, glycogenolysis via glucose-6-phosphatase, fatty acid elongation, lipolysis via hormone-sensitive lipase, calcium storage
|
|
|
intermediate filaments
|
10-12nm diameter; links extracellular matrix to cytoplasm and nucleus
|
|
|
microtubules
|
25nm diameter; consist of 13 circularly arranged a and b tubulins; associated with dynein (retrograde) and kinesin (anterograde) ATPases; polymerization inhibited by colchicine and vincristine; depolymerization inhibited by paclitaxel
|
|
|
cytokeratin
|
intermediate filament of epithelial cells
|
|
|
vimentin
|
intermediate filaments of endothelial and vascular smooth muscle cells, fibroblasts, chondroblasts and macrophages
|
|
|
desmin
|
intermediate filament of skeletal and non-vascular muscle
|
|
|
neurofilaments
|
intermediate filaments of neurons
|
|
|
glial fibrillar acidic protein
|
intermediate filaments of glia and microglia cells
|
|
|
cis regulators
|
core promoter sequence (TATA, BRE, DPE), proximal promoter region (GC box, CCAAT box), enhancers, silencers, insulators, response elements (CRE, SRE, IRE, GRE, PRE, HSRE, HMRE)
|
|
|
trans regulators
|
general transcription facotrs (TFIIs), gene regulatory proteins, CREB, serum response factor, Stat-1, Mep-1, AP1, steroid hormone receptor
|
|
|
homedomain proteins
|
helix-turn-helix with helix 3 being the DNA binding domain; OCT-1, OCT-2, Pit-1
|
|
|
leucine zipper proteins
|
alpha helix in which every 7th AA is leucine allows dimerization; CREB, Fos, Jun
|
|
|
helix-loop-helix proteins
|
two alpha helixes with a loop in between forms Y-shaped dimer; MyoD, Myc
|
|
|
transcription initiation complex
|
formed by RNA polymerase II and TFIIs at the core promoter TATA
|
|
|
serum response factor
|
trans factor binds SRE in response to serum growth factor
|
|
|
Stat-1
|
trans factor binds IRE in response to IFN-gamma
|
|
|
Mep-1
|
trans factor binds HMRE in response to heavy metals
|
|
|
AP1
|
trans factor binds PRE in response to phorbol esters
|
|
|
hsp70
|
trans binds HSE in response to heat shock
|
|
|
core promoter region
|
TATA interacts with RNA polymerase II and TFIIs
|
|
|
proximal promoter region
|
upstream from the core promoter; CCAAT box and GC-rich sequence
|
|
|
NFAT
|
transcription factor that increases expression of IL-2 gene; when T-cell receptor is enganged --> IP3 --> calcium activation of calcineurin phosphatase --> dephosphorylation of NFAT --> enters nucleus to upregulate transcription
|
|
|
Pit-1
|
gene regulatory homedomain protein required for transcription of GH, TSH and PRL; deficiency leads to pituitary dwarfism and hypopituitarism
|
|
|
miRNA
|
70bp inverted repeat that is cleaved by Dicer to form two interfering RNAs which blocks complimentary sequences of mRNA and prevents expression
|
|
|
alternative promoter
|
the first exon can vary with common downstream exons making different isoforms of same weight
|
|
|
alternative internal promoter
|
transcription is begun in at different promoters within the gene which produces proteins of different weight
|
|
|
alternative RNA splicing
|
spliceosome combines exons in different ways producinf different isoforms of the protein
|
|
|
lac operon: glucose+ lactose+
|
lac repressor is not bound to operator, CAP is not bound due to high cAMP --> lac operon is off
|
|
|
lac operon: glucose+ lactose-
|
lac repressor is bound to operator, CAP is not bound due to high cAMP --> lac operon is off
|
|
|
lac operon: glucose- lactose-
|
lac repressor is bound to operator, CAP is bound to due to low cAMP --> lac operon is off
|
|
|
lac operon: glucose- lactose+
|
lac repressor is not bound to operator, CAP is bound due to low cAMP --> lac operon is on
|