Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
55 Cards in this Set
- Front
- Back
- 3rd side (hint)
|
(1) What are the proportions of lipid-subtypes in the cell membrane?
(2) How does cholesterol effect the structure of a lipid membrane? |
(1)
50% phospholipids 50% cholesterol Traces of others |
(2)
Cholesterol stiffens and weakens the bilipid membrane because it interferes with the fluid interaction of phospholipids. |
|
|
Location of Vimentin
|
Intermediate filament found in connective tissue
|
|
|
|
Location of Desmin
|
Intermediate filament found in muscle
|
|
|
|
Location of Cytokeratin
|
Intermediate filament found in epithelial cells
|
|
|
|
Location of GFAP
|
Intermediate filament found in found in Neuroglia
|
|
|
|
Location of Neurofilaments
|
Intermediate filament found in neurons.
|
|
|
|
How does knowing the 5 intermediate filaments help us?
|
Because they're very large and abundant cytoskeletal structures, a stain specific for one of them will light up the tissue it lives in.
|
|
|
|
Ouabain
Mech: |
This toxin is found in leaves and bark in Africa and was used to poison arrow and spear-tips.
It blocks the K+ side of Na/K ATPase. |
|
|
|
Cardiac glycosides
Mech: |
Directly inhibit Na/K ATPase (slightly), decreasing the cells ability to pump out Ca2+.
Result: increased intracellular Ca2+ = increased cardiac contractility! |
|
|
|
Where are types II and III collagen found?
|
Type II collagen: found in cartilage.
Type III collagen: found in blood vessels, lymph nodes, dermis, and early wound healing. AKA reticulin |
|
|
|
Ehler Danlos?
Path & Presentation: |
Path: Type III collagen is defective d/t alpha chain mutation.
|
Presentation: joint hypermobility, hyperelastic skin, heart valve issues
|
|
|
Types of Collagen disorders:
|
Type I: Osteogenesis imperfecta, scurvy
Type III: Ehler Danlos Type IV: Alport's (defective) & Goodpastures (auto-antigenic) |
|
|
|
Describe Collagen Structure:
|
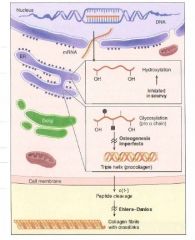
Triple helix made of Gly-X-Y repeats!
X and Y can be proline, hydroxyproline, or hydroxylysine! The Glycine residues line up axially, stabilizing the structure by their hydrophobic interactions. |
|
|
|
Steps of Collagen Synthesis:
|
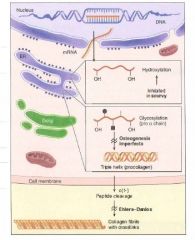
(1) Intracellular steps:
1. Translation of Gly-X-Y polypeptide. [preprocollagen] 2. Hydroxylation of lysin and proline residues (requires vitamin C) 3. Glycosylation of hydroxylysine residues |
(2) Extracellular steps:
4. Proteolytic processing: cleavage of terminal regions of procollagen [Tropocollagen, the basic monomer] 5. Tropocollagen strands link to eachother via lysine-hydroxylysine crosslinkages for enhanced strength. [Collagen fibril] |
|
|
Steps of Collagen Synthesis:
|
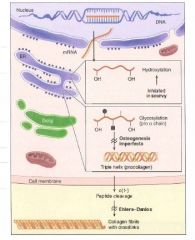
(1) Intracellular steps:
1. Translation of Gly-X-Y polypeptide. [preprocollagen] 2. Hydroxylation of lysin and proline residues (requires vitamin C) 3. Glycosylation of hydroxylysine residues |
(2) Extracellular steps:
4. Proteolytic processing: cleavage of terminal regions of procollagen [Tropocollagen, the basic monomer] 5. Tropocollagen strands link to eachother via lysine-hydroxylysine crosslinkages for enhanced strength. [Collagen fibril] |
|
|
How does intracellular collagen differ from extracellular collagen? Why must we finish making this stuff outside the cells?
|
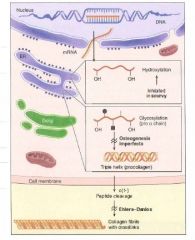
As pro- and preprocollagen, the molecule is still soluble.
Once the hydrophilic end segments are cleaved in the RER lumen, the molecule becomes insoluble, and would be lethal to cells. |
|
|
|
What are the most common and most deadly forms of osteogenesis imperfecta? How do they differ?
|
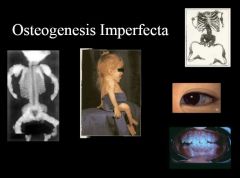
Osteogenesis Imperfecta Type I:
Mildest form Autosomal dominant Osteogenesis Imperfecta Type II: autosomal recessive Lethal, perinatally |
|
|
|
Steps of PCR:
|
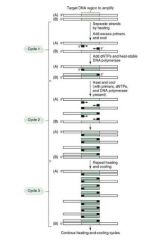
1. Denaturation:
Add lots of mRNA primers, dNTPs, and heat-stabile DNA Polymerase from geyser bacterium to your sample, and heat to denature it. 2. Reanneal: cool enough for RNA primers to anneal, and for special DNA-P to do its work. Repeat, repeat, repeat |
Concept: we use RNA primers that we know to be flanking the gene we want to amplify.
After several cycles, we'll have tons of DNA from that gene only. |
|
|
How do we visualize our PCR results?
|
This lab test is visualized using gel electrophoresis on agarose gel.
Shorter pieces of DNA travel further. We then compare the gel against a DNA ladder. |
|
|
|
Which Blotting Technique does what?
|
SNoW
DRoP Southern Blot: used to separate DNA Northern Blot: used to separate RNA Western Blot: used to separate Protein |
|
|
|
Materials for a Laboratory Blot:
|
1. DNA/RNA/Protein is electrophoresed on agarose gel, then transferred to a filter.
2. Denaturant is applied and radiolabeled primers are added [nucleic acid primers for Noorthern and Southern blots, and antibody primers for Western blot] 3. Read by exposing to film |
|
|
|
Laboratory Blot Techniques:
|
1. DNA/RNA/Protein is electrophoresed on agarose gel, then transferred to a filter.
2. Denaturant is applied and radiolabeled primers are added [nucleic acid primers for Noorthern and Southern blots, and antibody primers for Western blot] 3. Read by exposing to film |
|
|
|
How does microarray work?
|
We immobilize thousands of DNA fragments from known genes on a chip, then add radiolabeled primers from our patient sample.
I have no idea how we get radiolabeled primers from a patients blood/CSF/urine sample, but we do. |
|
|
|
How do ELISA's work?
|
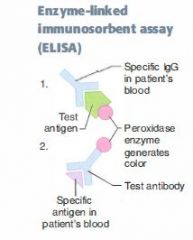
If we're testing for the presence of an antibody in the patient's serum, add its antigen with peroxidases specially attached.
If we're testing for presence of an antigen in the patient's serum, add specific antibodies with peroxidases attached to the sample. Either way, if the patient is sero-positive, there'll be an intense color reaction. |
|
|
|
Pleiotropy
|
1 gene has more than 1 effect on an individual's phenotype.
i.e. PKU: it's a monogenic disorder, but causes MR, hair and skin changes, and a bunch of other seemingly unrelated problems. |
|
|
|
Loss of heterozygosity
|
The 2nd hit of the 2-hit hypothesis.
|
|
|
|
Dominant negative mutation
Give an example |
When a heterozygote produces 1 mutant allele that doesn't only fail to work, but also interferes with the function of the other, normal allele.
|
Example: a mutation of a Transcription factor allele in its allosteric site: allele can still bind its target, blocking out the good copy of the TF from acting!
|
|
|
Lyonization
|
Random X inaction in females, resulting in one normal X chromosome and one Barr body.
Perfectly normal, but it means that any mutations on the normal X chromosome will be dominant. |
|
|
|
Locus heterogeneity
|
When mutations at two different loci cause the same phenotype.
|
Occurs in Marfan's, MEN 2B, and homocystinuria, all of which result in clinical marfan's syndrome.
|
|
|
Heteroplasma
|
Inheritance of both normal and mutant mitochondrial DNA (mtDNA), resulting in variable expression of mitochondrial disease.
|
Depends upon which mitochondria were inherited by the lucky germ cell!
|
|
|
Achondroplasia
Etiology and biochem Inheritance pattern |
FGFR3 (fibroblast growth factor 3) defect resulting in dwarfism, short limbs, but head and trunk are normal size.
Autosomal-dominant |
Autosomal Dominant
|
|
|
Autosomal Dominant Polycystic Kidney Disease (ADPKD)
[the adult form] Etiology: |
Always bilateral, massive enlargement of kidneys d/t multiple large cysts.
Presents with flank pain, flank swelling, hematuria. |
Autosomal dominant
**Associated with berry aneurisms An infantile recessive form exists that's much worse. |
|
|
Familial Hypercholesterolemia
|
Elevated LDL d/t defective or absent LDL receptor.
Presents with MI, very hi cholesterol. Homozygotes are rare and don't live long. |
Autosomal Dominant
|
|
|
Familial Adenomatous Polyposis
|
Colon becomes covered in polyps. Colorectal cancer is inevitable.
APC mutation on chromosome 5. [5 letters in polyp] |
Autosomal Dominant
|
|
|
Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome)
Etiology: Pattern of Inheritance: |
Inherited disorder of blood vessels. Findings include telangiectasia, recurrent epistaxis, arteriovenous malformations.
Presents with purpura, nosebleeds, telangiectasia on tongue. |
Autosomal Dominant
|
|
|
Hereditary spherocytosis
Etiology: Pattern of Inheritance: |
Spheroid eryhthrocytes d/t ankyrin or spectrin defect.
Presents with hemolytic anemia, increased MCHC. This is the disorder in which the wire mesh of the normal spleen is responsible for your hemolysis. |
Autosomal Dominant
|
|
|
Marfan's Syndrome
Etiology: Pattern of Inheritance: |
Fibrillin gene mutation resulting in connective tissue abnormalities.
|
Autosomal Dominant
|
|
|
MEN
Etiology: Pattern of Inheritance: |
Familial tumors of the endocrine glands.
Types 2A and 2B are caused by RET mutation. |
All 3 syndromes are autosomal dominant
|
|
|
Neurofibromatisis type 1
Etiology: Pattern of Inheritance: |
Cafe Au lait spots
neural tumors blindness Lisch nodules are ubiquitous Mutation on long arm of chromosome 17 |
Autosomal Dominant
|
|
|
Neurofibromatosis type 2
|
Bilateral acoustic schwannomas
Juvenile cataracts NF2 on Chromosome 22 [2's disease: Type 2 on chrom. 22] |
Autosomal dominant
|
|
|
Tuberous sclerosis
|
Facial lesions
Hamartomatous masses in cortex and retina Increased incidence of astrocytomas |
Autosomal dominant
|
|
|
von Hippel-Lindau disease
|
Hemangioblastomas of retina/cerebellum/medulla.
Half of these individuals also develop renal cell carcinoma. Familial form is caused by VHL gene mutation on p arm of chromosome 3. |
Autosomal dominant
There is also a sporadic form caused by loss of p arm of chromosome 3. |
|
|
VHL
(1) Genetics & biomolecular changes (2) Path |
(1)
Gene located on 3p. Can be lost d/t del(3p) or mutation to the VHL gene on 3p. VHL normally degrades the transcription factor HIF-1α, preventing it from triggering PDGF and VEG-2F expression. |
Disease consists of medulloblastomas, hemangiomas of the retina/cerebellum/medulla.
Half of these individuals also will get renal cell carcinoma. Bottom line: when VHL is knocked out, we'll see cancerous expansion of many small blood vessels. |
|
|
Cystic Fibrosis
Normal vs. pathological pulmonary biochem: |
Normal Lungs:
CFTR is a Cl- channel that actively secretes chloride in lungs CFTR regulates ENaC, preventing excessive Na+ absorption by the lung epithelium. |
Lungs in CF:
1. Cl- channel is absent or defective, so less Cl- is secreted from lung epithelium. 2. Cl- channel cannot, under these circumstances, downregulate ENaC activity, and Na+ is reabsorbed into lung epithelium too vigorously 3. H2O follows Na+ out of the airways and into the epithelium 4. Result: thick, stagnant mucous in airways that forms plugs and invites bacterial colonization. so it can't regulate ENaC, and lots of Na+ (and therefore H2O) is reabsorbed. |
|
|
Genetics of CF & Location of Cystic Fibrosis gene
|
CFTR is located on the long arm of Chromosome 7
Autosomal recessive inheritance pattern |
|
|
|
Complications of Cystic Fibrosis
|
Lung:
1. recurrent infections by pseudomonas and staph aureaus 2. chronic bronchitis, bronchiectasis GI: pancreatic insufficiency |
|
|
|
Compare CF pathophys in skin vs. lung
|
In lung, CF is caused by defect in a Cl- channel that normally actively SECRETES Cl-.
In the skin, CF is caused by defect in a Cl- channel that actively REABSORBS Cl- In both cases, pathological buildup of Cl- is mirrored by pathological buildup of Na+ and H2O, because CFTR regulates ENaC. |
|
|
|
Cystic fibrosis
Normal vs. pathological biochem in skin: |
Normal Skin:
1. CFTR is a Cl- transporter that actively *reabsorbs* Cl- from sweat 2. CFTR upregulates ENaC activity, and therefore Na+ reabsorption. |
Skin in CF:
1. CFRT is absent or defective, so less Cl- is reabsorbed by the skin. 2. Cl- channel cannot regulate ENaC, so Na+ isn't reabsorbed either. 3. Lots of NaCl is lost in sweat. |
|
|
Treatment of CF
|
Palliative:
1. Give N-Acetylcysteine to loosen mucous plugs 2. Give lots of electrolytes. LOTS! Gene modification therapy is in the works but not yet ready. |
|
|
|
List the X-Linked Recessive disorders
|
B W F G O L D H H
(Be Wise, Fool's GOLD Heeds Silly Hopes) Bruton's aggamaglobulinemia Wiskott-Aldrich syndrome Fabry's Disease G6PD Deficiency Ocular Albinism Lesch-Nyhan syndrome Duchenne's muscular dystrophy Hunter's Syndrome Hemophilia A and B |
|
|
|
Duchenne's Muscular dystrophy
Genetics & Biochem |
(1)
X-linked Recessive Deletion/frameshift mutations in Dystrophin protein |
(2) Biochem:
Dystrophin connects Actin of cytoskeleton to sarcolemmal membrane of myocytes. Result of deletion: force of muscle fiber contraction is not transferred to cell membrane; the cell doesn't contract, only its organelle does. |
|
|
Becker's Muscular Dystrophy
Genetics & Biochem |
(1)
X-linked recessive Missense/nonsense Mutation (*NOT* frameshift/deletion) to dystrophin gene. |
(2) Biochem:
Some dystrophin is made, but it's abnormal to a greater or lesser degree. The result is a disorder that's less severe and has a later onset than in Duchenne's. |
|
|
Muscular dystrophies
Presentation & Diagnostics |
These disorders are inherited and present at anytime from childhood to early adulthood wiht progressive muscle wasting and myocyte degredation.
Histological abnormalities include: Muscle tissue can become replaced with fat. Muscle fibers will vary in size and diameter. |
Diagnose via increased serum Creatinine Kinase and muscle biopsy.
|
|
|
Fragile X
Path & Presentation |
X-linked defect in methylation and expression of the FMRI gene.
Results in trinucleotide expansion repeat of CGG |
Second most common cause of MR!
X-linked, X-tra long gene, Xtra large testes, jaw, and ears!! |
|
|
List the trinucleotide repeat diseases and their repeated sequences.
|
1. Fragile X: CCG
2. Friedrich's ataxia: GAA 3. Huntington's Disease: CAG 4. Myotonic Dystrophy: CTG |
|