Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
84 Cards in this Set
- Front
- Back
|
What part of hemostasis are platelets involved in?
|
Primary hemostasis
|
|
|
What are the characteristics of the structure/contents of platelets?
|
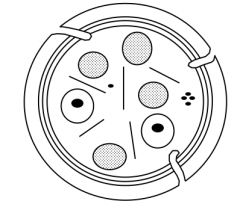
- Anucleate cells
- Plasma membrane contains proteins that provide platelets w/ unique characteristics - Granules / vesicles inside cell (2 types - gray and one that contains e- dense core = black circle) - Marginal rim of microtubules - Membrane invaginates to form internal membrane system that makes its way through |
|
|
What do platelets do when there is damage to the endothelium?
|
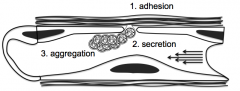
1. ADHESION of platelets to subendothelium through damaged endothelium
2. SECRETION of granules through surface connected cannaliculus system, which attracts other platelets to the area 3. AGGREGATION of platelets to initiate primary hemostasis to cover defect |
|
|
How do platelets mediate binding to the subendothelial structures after damage to endothelium?
|
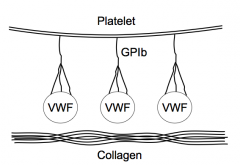
- Platelets have VWF on their surface attached via GPIb protein
- VWF then binds to collagen - There are other integrins that can mediate binding to subendothelial structures too |
|
|
What happens when platelets are activated?
|
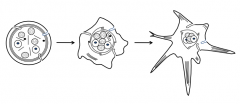
Platelets receive biochemical signal to change shape
- Originally round → irregular → pseudopods sticking out - Pseudopods make contact with subendothelial CT and other platelets - Marginal rim of microtubules and contractile proteins are activated by increased Ca2+ |
|
|
What are the types of platelet storage granules?
|
- Dense (delta) granules
- Alpha granules - Lysosomal (lambda) granules |
|
|
What is found in the dense (delta) granules?
|
- Nucleotides: ADP and ATP
- Serotonin (vasoactive amine) - Calcium |
|
|
What is found in the alpha granules?
|
- Fibrinogen
- Factor V - Thrombospondin - PDGF, PF4, βTG (platelet growth factors) |
|
|
What is the action of nucleotides (ADP) on platelets?
|
Agonist for platelets, causes aggregation
|
|
|
What is the action of Phospholipase A in the membrane of platelets?
|
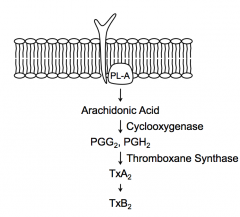
Cleaves phospholipids in membrane to generate Arachidonic Acid in cytoplasm
|
|
|
What happens to Arachidonic Acid in cytoplasm of platelets after being generated by Phospholipase A in the membrane?
|
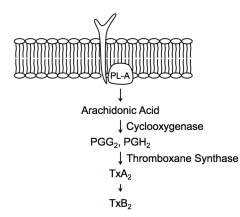
- Cyclooxygenase converts AA → PGG2 and PGH2
- Thromboxane Synthease converts PGG2 and PGH2 → TxA2 - Spontaneously, TxA2 converts to TxB2 |
|
|
What expresses Thromboxane Synthase? Action?
|
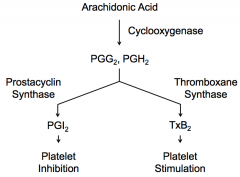
Platelets express Thromboxane Synthase, which converts PGG2 and PGH2 → TxA2 → spontaneously converts to TxB2 → stimulates platelets
|
|
|
What expresses Prostacyclin Synthase? Action?
|
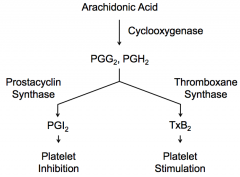
Endothelial cells express Prostacyclin Synthase, which converts PGG2 and PGH2 → PGI2 → inhibits platelets from sticking to each other
|
|
|
What is necessary to prevent platelets from aggregating when there is no endothelial injury?
|
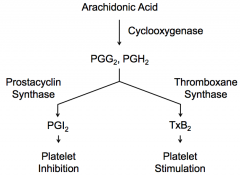
Endothelial cells express Prostacyclin Synthase, which converts PGG2 and PGH2 → PGI2 → inhibits platelets from sticking to each other
|
|
|
How do platelets mediate aggregation
|
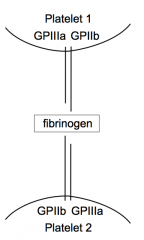
- Platelets express GPIIIa and GPIIb
- These receptors bind Fibrinogen - Fibrinogen binds to other GPIIIa and GPIIb receptors on other platelets binding the two together leading to aggregation |
|
|
What are the types of platelet disorders?
|
Quantitative:
- Decreased platelet count - Prolonged bleeding time Qualitative: - Normal platelet count - Prolonged bleeding time (can have a mixture of both types of defects) |
|
|
What are the causes of defects of platelet adhesion (qualitative defect)?
|
- Von Willebrand's Disease (cofactor abnormality)
- Bernard-Soulier Syndrome (platelet abnormality) - Ehlers Danlos Syndrome (substrate abnormality) - Scurvy (substrate abnormality) - Pseudoxanthoma Elasticum (substrate abnormality) |
|
|
What are the causes of defects of platelet secretion (qualitative defect)?
|
Prostaglandin defects:
- Phospholipase defect - Cyclooxygenase defect (aspirin and NSAIDs) - Thromboxane synthase defect Storage pool defects (SPD): - Alpha SPD - Delta SPD - Alpha-Delta SPD |
|
|
What are the causes of defects of platelet aggregation (qualitative defect)?
|
- Glanzmann's Thrombasthenia (membrane changes)
- Uremia (abnormal milieu) - Fibrin split productions (abnormal milieu) |
|
|
What are the causes of defects of platelets due to increased utilization (quantitative defect)?
|
- Diffuse Intravascular Coagulation
- Thrombotic Thrombocytopenic Purpura - Abnormal Heart Valves |
|
|
What are the causes of defects of platelets due to increased destruction (quantitative defect)?
|
- Idiopathic Thrombocytopenic Purpura (more common)
- Lupus Erythematosus (Abs against platelets) - Drugs: Quinidine, and many others (cause development of Abs against platelets) |
|
|
What are the two forms of Idiopathic Thrombocytopenic Purpura (ITP)?
|
- Acute ITP
- Chronic ITP |
|
|
What are the signs of Acute vs Chronic ITP?
|
- Acute ITP: prominent bleeding and purpura, no prior bleeding history
- Chronic ITP: bleeding less prominent, long history of easy bruising, indolent |
|
|
Who is typically affected by Acute vs Chronic ITP?
|
- Acute: usually children (M:F ~1:1), frequently preceded by a viral syndrome
- Chronic: usually adults (M:F ~ 1:3) |
|
|
What are the characteristics of ITP antibodies?
|
- Platelet specific
- Restricted heterogeneity (IgG3 - κ and λ light chains) - Antigenic heterogeneity (GPIb, GPIIb, or GPIIIa) - Site of production of Abs: spleen and/or BM |
|
|
What kind of antibodies are there in ITP?
|
- IgG3
- κ and λ light chains |
|
|
What do ITP antibodies target?
|
Usually target surface proteins on platelets: GPIb, GPIIb, or GPIIIa
(remember IgG!) |
|
|
How do you diagnose ITP?
|
Diagnosis of exclusion
- Must be evidence of immune thrombocytolysis (thrombocytopenia, normal or increased megakaryocytes, anti-platelet antibody) - Absence of other causes of thrombocytopenia (lupus erythematosus, diffuse intravascular coagulation, drugs, lymphoma) - Absence of splenomegaly |
|
|
What is the evidence for immune thrombocytolysis to make the diagnosis of ITP?
|
- Thrombocytopenia (low platelet count)
- Normal or increased megakaryocytes (if these were low, then it is a production problem rather than a destruction problem) - Anti-platelet antibody |
|
|
What other causes of thrombocytopenia must you rule out to make the diagnosis of ITP?
|
- Lupus Erythematosus
- Diffuse Intravascular Coagulation - Drugs - Lymphoma |
|
|
There must be an absence of what physical exam feature to make a diagnosis of ITP? Why?
|
Absence of splenomegaly - if spleen is enlarged it could be the cause of the thrombocytopenia
|
|
|
How do you treat ITP?
|
- Corticosteroids and IVIG
- Splenectomy (if corticosteroids aren't working) - Immunosuppressive agents |
|
|
How effective are corticosteroids for treating ITP? Effects?
|
- 70% response with high relapse rate
- Decreases antigen processing and antibody production |
|
|
How effective is a splenectomy for treating ITP? Effects?
|
- 50% response with low relapse rate
- Removes antibody source and site of sequestration - Must give Pneumovax to children (because they will be at increased risk for infections) |
|
Case 1:
- 18 yo female presents for evaluation of red rash on her bilateral ankles - Progressively increased over 3 days - First notice after playing v-ball - No fever, chills, lymphadenopathy, recent illness or meds - Last menstrual period was 3 weeks ago w/ moderate bleeding (5 days, no clots) - Exam: petechiae on palate, rash on bilateral legs extending to knee - WBC normal, Hemoglobin normal, platelet count low (6000) - PT, INR, aPTT, and fibrinogen all normal What is the most appropriate diagnostic study? a) Platelet function assay b) Peripheral blood smear c) von Willebrand’s activity test d) Rheumatoid factor e) Factor VIII activity |
Peripheral blood smear
When you are given a choice in hematology about what test to do, it is almost always peripheral blood smear. We want to know what is driving this thrombocytopenic condition. Are there leukemia cell? Are there other problems with RBC fragmentation? Other suggestions of platelet system being activated? |
|
|
Case 1:
- 18 yo female presents for evaluation of red rash on her bilateral ankles - Progressively increased over 3 days - First notice after playing v-ball - No fever, chills, lymphadenopathy, recent illness or meds - Last menstrual period was 3 weeks ago w/ moderate bleeding (5 days, no clots) - Exam: petechiae on palate, rash on bilateral legs extending to knee - WBC normal, Hemoglobin normal, platelet count low (6000) - PT, INR, aPTT, and fibrinogen all normal What does our differential diagnosis include? |
- Idiopathic Thrombocytopenic Purpura (ITP)
- Lupus Erythematosus - Diffuse Intravascular Coagulation (no other lab abnormalities) - Drugs (not taking any) - Lymphoma (rule out with peripheral blood smear) |
|
|
Case 1:
- 18 yo female presents for evaluation of red rash on her bilateral ankles - Progressively increased over 3 days - First notice after playing v-ball - No fever, chills, lymphadenopathy, recent illness or meds - Last menstrual period was 3 weeks ago w/ moderate bleeding (5 days, no clots) - Exam: petechiae on palate, rash on bilateral legs extending to knee - WBC normal, Hemoglobin normal, platelet count low (6000) - PT, INR, aPTT, and fibrinogen all normal Patient is diagnosed with ITP. How should she be treated? |
Treatment with Prednisone and IVIG
- Her platelet counts respond to normal levels and the petechiae resolves - No further bleeding complications |
|
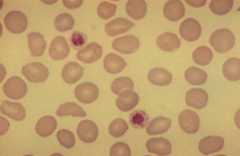
What are the characteristics of these platelets?
|
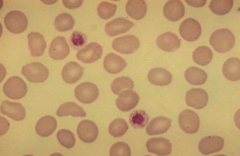
- They are large relative to RBCs (about the same size)
- We can see these platelets have granules (taken up dye) |
|
Case 1:
- Girl diagnosed with ITP - Does well for 10 years without bleeding complications - Presents for evaluation of fatigue - She is a runner, but reports decreased exercise tolerance and loss of pace - Current meds: aspirin for ankle pain and ibuprofen for menstrual cramps - Vegetarian but eats fish and eggs - Increased menorrhagia over past 2 years - Exam: conjunctival pallor, mild tachycardia (100bpm), late systolic murmur, multiple bruises on bilateral upper extremities - Labs: WBC and platelet counts normal; hemoglobin LOW and MCV LOW; PT, INR, aPTT, and fibrinogen normal What is the most likely cause of this patient’s anemia? a) Acute blood loss b) Iron deficiency anemia c) Vitamin B12 deficiency d) Hemolytic anemia e) Beta thalassemia minor |
Iron deficiency anemia (vegetarian, menorrhagia, aspirin & NSAIDs)
|
|
|
Case 1:
- Girl diagnosed with ITP - Does well for 10 years without bleeding complications - Presents for evaluation of fatigue - She is a runner, but reports decreased exercise tolerance and loss of pace - Current meds: aspirin for ankle pain and ibuprofen for menstrual cramps - Vegetarian but eats fish and eggs - Increased menorrhagia over past 2 years - Exam: conjunctival pallor, mild tachycardia (100bpm), late systolic murmur, multiple bruises on bilateral upper extremities - Labs: WBC and platelet counts normal; hemoglobin LOW and MCV LOW; PT, INR, aPTT, and fibrinogen normal What would you do to confirm diagnosis of iron deficiency anemia? |
- Ferritin
- Iron (transferrin) saturation - Total iron binding capacity - Reticulocyte count |
|
|
What are the causes of microcytic anemias?
|
- Iron deficiency
- Anemia of chronic disease (inflammation) - Thalassemias - Sideroblastic anemia |
|
|
What type of anemia:
- ↓ MCV - ↑ RDW (RBC Distribution Width) - ↓ Iron Sat% - ↓↓ Ferritin - ↓↓ BM Iron |
Iron Deficiency Anemia
|
|
|
What type of anemia:
- ↓/Nl MCV - Nl RDW (RBC Distribution Width) - ↓/Nl Iron Sat% - Nl/↑ Ferritin - ↑ BM Iron |
Anemia of Chronic Disease (inflammation)
|
|
|
What type of anemia:
- ↓ MCV - Nl/↑ RDW (RBC Distribution Width) - ↑ Iron Sat% - ↑↑ Ferritin - ↑↑ BM Iron |
Thalassemias
|
|
|
What type of anemia:
- ↓ MCV - ↑ RDW (RBC Distribution Width) - Nl/↑ Iron Sat% - ↑ Ferritin - ↑ BM Iron |
Sideroblastic Anemia
|
|
|
Case 1:
- Girl diagnosed with ITP, returns 10 years later w/ complaints of fatigue - Labs: WBC and platelet counts normal; hemoglobin LOW and MCV LOW; PT, INR, aPTT, and fibrinogen normal - Von Willebrand's antigen, activity, and multimers normal - Factor VIII activity normal - Thrombin time normal - Platelet function assay: prolongation on collagen/epi cartridge (>300s, Nl <180s); normal on collagen/ADP cartridge What is the mechanism of the patient’s platelet function defect? a) impaired platelet adhesion to endothelial damage due to impaired von Willebrand's factor collagen binding b) quantitative defect in the number of platelets available for coagulation c) decreased thrombin generation due to suppression of clotting factors d) decreased platelet aggregation due to reduced formation of thromboxane A2 e) development of a factor inhibitor (autoantibody) against factor VIII |
Decreased platelet aggregation due to reduced formation of thromboxane A2
This is happening because the patient is on aspirin |
|
|
What happens in a Platelet Function Assay?
|
- Take tube that has blood in it
- Platelets exposed to agonists that cause blood to clot (eg, collagen/Epi, collagen/ADP) - Not very specific - Epi and ADP promotes clotting |
|
|
If a patient's platelet function assay shows prolongation on collagen/epi cartridge (>300s, Nl <180s) but a normal on time for the collagen/ADP cartridge, what is your differential? How do you determine which is right?
|
- Drugs (check what drugs they are on)
- Von Willebrand Factor problem (run von Willebrand studies) - ITP (diagnosis of exclusion) |
|
|
What are the effects of Aspirin on platelets?
|
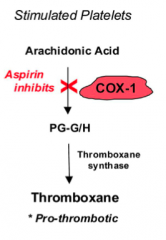
- Decreases platelet aggregation by inhibiting COX-1
- Results in decreased thromboxane formation - Thromboxane regulates platelet aggregation through mediating glycoprotein IIb/IIIa |
|
|
What is the function of primary hemostasis? What happens if there is a problem with it?
|
- Primary hemostasis → formation pf platelet plug
- Problems → mucocutaneous bleeding, including petechiae and mucosal bleeding (epistaxis, gingival bleeding, menorrhagia) |
|
|
What are the types of disorders that affect primary hemostasis?
|
- Qualitative and quantitative platelet defects
- vW disease - Disorders of endothelium |
|
|
What initiates the extrinsic clotting cascade?
|
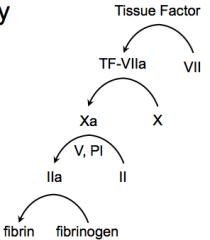
Initiated by things that are outside of the blood and endothelium = Tissue Factor
|
|
|
What initiates the intrinsic clotting cascade?
|
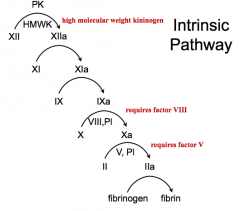
Things in the blood = Factor XII and PK
|
|
|
What is the function of the extrinsic vs intrinsic clotting cascade?
|
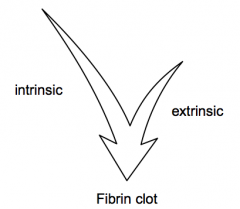
- Extrinsic cascade gets coagulation started
- Intrinsic cascade links to inflammatory system and helps keep clotting going |
|
|
What is the final result of the coagulation via the intrinsic or extrinsic pathway?
|
Fibrin clot - which occurs in and among platelets, leading to stopped bleeding, and wound healing
|
|
|
What happens in the extrinsic pathway?
|
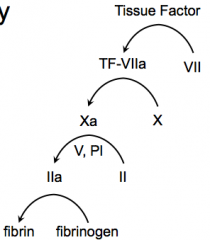
- Tissue Factor activates Factor VII → TF-VIIa
- TF-VIIa activates Factor X → Xa - Xa activates Prothrombin (II) → IIa with help from V, and PI - IIa activates Fibrinogen → Fibrin |
|
|
What factors are involved in the Extrinsic Pathway?
|
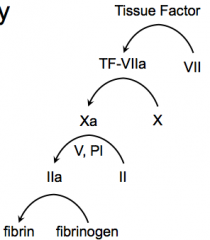
TF, 7, 10, 2, Fibrin
- Tissue Factor - Factor VII - Factor X - Factor II (Prothrombin) w/ help from Factor V and PI - Fibrinogen |
|
|
What happens in the intrinsic pathway?
|
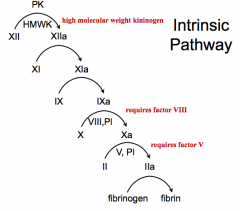
- PK and HMWK activate XII → XIIa
- XIIa activates XI → XIa - XIa activates IX → IXa - IXa activates X → Xa (requires factor VIII, PI) - Xa activates Prothrombin (II) → IIa (requires factor V, PI) - IIa activates Fibrinogen → Fibrin |
|
|
What factors are involved in the Intrinsic Pathway?
|
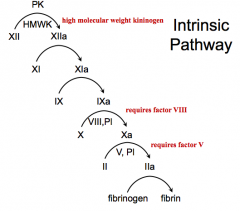
PK + 12, 11, 9, 10 (requires 8), 2 (requires 5), Fibrin
- PK + Factor XII - Factor XI - Factor IX - Factor X (requires factor VIII) - Factor II (requires factor V) - Fibrinogen |
|
|
What is the role of Vitamin K in the clotting cascade?
|
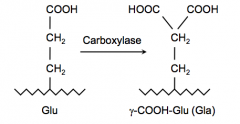
Helps convert glutamic acid to γ-carboxy-glutamic acid via Carboxylase
γ-carboxy group helps these factors to bind Ca2+ which is necessary for the conformational changes to help these factors be activated |
|
|
Which factors and other proteins require Vitamin K?
|
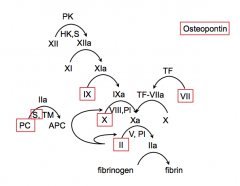
- Factors II, VII, IX, X (2, 7, 9, 10)
- Proteins: PC, S, Osteopontin |
|
|
What does the Prothrombin Time (PT) test?
|
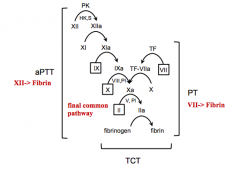
Extrinsic coagulation
(VII) |
|
|
What does the Activated Partial Thromboplastin Time (aPTT) test?
|
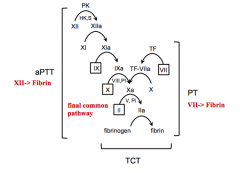
Intrinsic coagulation
(XII, XI, IX, VIII) |
|
|
What does the Thrombin Clotting Time (TCT) test assess?
|
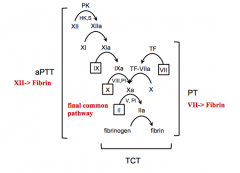
Ability of fibrinogen to convert to fibrin in final common pathway
|
|
|
What can cause an isolated prolonged Activated Partial Thromboplastin Time (aPTT)?
|
Helps assess the intrinsic pathway:
- High Molecular Weight Kininogen deficiency - Prekallikrein deficiency - F XII deficiency - F XI deficiency - F IX deficiency - F VIII deficiency - Passovoy Deficiency - Lupus anticoagulant |
|
|
How can you determine if there are antibodies to factors causing a deficiency or if there is just a genetic deficiency of a factor?
|
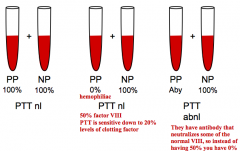
Mixing Studies for Inhibitor:
- Only do this test if there is an abnormal PTT or PT to identify it due to antibodies |
|
|
If you mix a hemophiliac's blood (deficiency of factor) with a normal patient's blood, what will the results be for the PTT or PT?
|
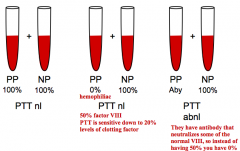
The PTT or PT will be normal because there will be 50% of that factor, and the test only detects deficiency if <20% levels of the clotting factor
|
|
|
If you mix a patient with antibody to a certain factor with a normal patient's blood, what will the results be for the PTT or PT?
|
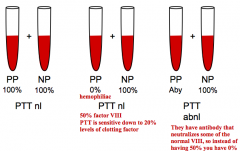
PTT or PT will still be low because the normal factor from the normal patient will be neutralized by the antibody
|
|
|
What can cause an isolated prolonged PT?
|
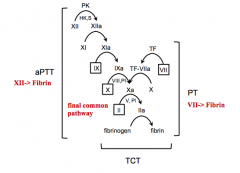
Factor VII deficiency
|
|
|
What can cause a prolonged PT and aPTT?
|
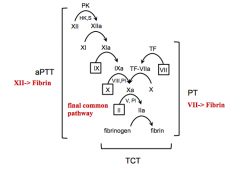
Parts of final common pathway
- Factor X deficiency - Factor V deficiency - Factor II deficiency - Fibrinogen deficiency |
|
|
What can cause an isolated prolonged TCT?
|
- Afibrinogenemia
- Hypofibrinogenemia - Hyperfibrinogenemia - Dysfibrinogenemia - FDPs - Immunoglobulins - Heparin |
|
|
What can cause bleeding if the screening tests come back normal?
|
- Mild factor deficiency
- Thrombocytopenia - Thrombocytopathia - Vascular abnormality (scurvy) - Factor XIII deficiency - Antiplasmin deficiency |
|
|
Case 2:
- 5 yo boy seen post-anesthesia with post-operative bleeding - He has been bleeding for 3 hrs after tonsils and adenoidectomy - Spitting up blood without cough - Intraoperatively, procedure was uneventful with mildly increased diffuse bleeding - PMH: normal circumcision, recurrent sore throats treated w/ antibiotics, otherwise helathy - Meds: clindamycin, ibuprofen, dexamethasone, fentanyl, propofol intraoperatively - Normal family hx and social hx - No history of bleeding or bruising symptoms - Exam: no rashes or bruises - Labs: all normal, except elevated PTT (41 sec) What is the next most appropriate diagnostic step? a) von Willebrand’s activity testing b) Fibrinogen c) Partial thromboplastintime 1:1 mixing study d) Platelet function assay e) D-dimer |
Partial thromboplastintime 1:1 mixing study
Is there a factor deficiency or is there an inhibitor / antibody that is prolonging the clotting time in the test tube? PTT mixing study is very fast so you should get the results very quickly |
|
|
Case 2:
- 5 yo boy seen post-anesthesia with post-operative bleeding - He has been bleeding for 3 hrs after tonsils and adenoidectomy - Spitting up blood without cough - Intraoperatively, procedure was uneventful with mildly increased diffuse bleeding - PMH: normal circumcision, recurrent sore throats treated w/ antibiotics, otherwise helathy - Meds: clindamycin, ibuprofen, dexamethasone, fentanyl, propofol intraoperatively - Normal family hx and social hx - No history of bleeding or bruising symptoms - Exam: no rashes or bruises - Labs: all normal, except elevated PTT (41 sec) What is the next appropriate management step? |
- Assessment of patient’s airway with intubation if required
- IV fluids - Type and cross for blood transfusion - Surgical hemostasis - Further lab studies to continue making diagnosis |
|
|
Case 2:
- 5 yo boy seen post-anesthesia with post-operative bleeding - He has been bleeding for 3 hrs after tonsils and adenoidectomy - Spitting up blood without cough - Intraoperatively, procedure was uneventful with mildly increased diffuse bleeding - PMH: normal circumcision, recurrent sore throats treated w/ antibiotics, otherwise helathy - Meds: clindamycin, ibuprofen, dexamethasone, fentanyl, propofol intraoperatively - Normal family hx and social hx - No history of bleeding or bruising symptoms - Exam: no rashes or bruises - Labs: all normal, except elevated PTT (41 sec) - Patient returns to OR and hemostasis achieved w/ cautery, no evidence of trauma or lacerated vessels - Labs: PTT still elevated and PTT 1:1 mix is corrected What lab result would best explain the patient’s bleeding complications? a) Lupus anticoagulant positive b) ↓ factor VIII activity c) Loss of highest weight von Willebrand’s multimers d) ↓ fibrinogen e) ↓ factor VII activity |
↓ factor VIII activity
There is a deficiency of one of the intrinsic pathway factors Factor VII deficiency would be if it was a prolonged PT but normal PTT |
|
|
Case 2:
- 5 yo boy seen post-anesthesia with post-operative bleeding - He has been bleeding for 3 hrs after tonsils and adenoidectomy - Spitting up blood without cough - Intraoperatively, procedure was uneventful with mildly increased diffuse bleeding - PMH: normal circumcision, recurrent sore throats treated w/ antibiotics, otherwise helathy - Meds: clindamycin, ibuprofen, dexamethasone, fentanyl, propofol intraoperatively - Normal family hx and social hx - No history of bleeding or bruising symptoms - Exam: no rashes or bruises * Labs: PTT still elevated and PTT 1:1 mix is corrected * Factor VIII activity ↓ (13%), Factor IX nl (88%), Factor XI nl (125%), Factor XII nl (105%) What is the appropriate diagnosis? |
Mild Hemophilia A
|
|
|
What is a Hemophilia?
|
Inherited bleeding disorder in which there is a deficiency or lack of Factor VIII (hemophilia A) or Factor IX (hemophilia B)
|
|
|
Which factors are deficient in Hemophilia A and B?
|
- Hemophilia A: deficient Factor VIII
- Hemophilia B: deficient Factor IX |
|
|
How do you get hemophilia?
|
- 70% of cases are inherited (X-linked recessive)
- Typically expressed in males but carried in females - Severity consistent between family members - 30% of cases are new mutations |
|
|
What are the degrees of Hemophilia severity?
|
- Mild hemophilia: 6-50%
- Moderate hemophilia: 1-5% - Severe hemophilia: <1% |
|
|
What is the pattern of bleeding most commonly identified in hemophilia?
|
- Ooozing - can’t stabilize the fibrin clot
- Late re-bleeding - Articular bleeds (joints, soft tissues) - more common in defects of secondary hemostasis |
|
|
How should a patient with Hemophilia be treated?
|
Treated with recombinant factor VIII (hemophilia A) or factor IX (hemophilia B)
|
|
|
In a hemophilic patient who is given the recombinant factor they are missing, what could cause the factor activity to go down leading to bleeding?
|
Development of an inhibitor (antibody) against that factor (this is the most serious complication of factor replacement therapy)
|
|
|
How should you treat a patient with an inhibitor / antibody to a factor?
|
- Polyclonal high affinity IgG Ab that neutralize the procoagulant function of fVIII or fIX
- Factor replacement therapy becomes ineffective unless antibody is removed |