Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
196 Cards in this Set
- Front
- Back
|
Definition of Edocrinology
|
cellular communication by means of hormones
|
|
|
hormone
|
chemical messenger released by an endocrine (ductless) gland, transported in low concentrations to target cells
|
|
|
neurohormone
|
chemical messenger secreted by neurons that is transported via circulation to target cells
|
|
|
receptors
|
determine the “target” cell and the specific action of hormone; either expressed on cell surface or in cytoplasm/nucleus
|
|
|
Endocrine organs and their endocrine function
|
• thyroid - metabolism
• parathyroid - calcium metabolism • adrenal - lots of stuff that affects salt and water metabolism, stress • kidney - Vit D conversion • gonads - puberty, reproduction • pituitary - master regulator, controls many peripheral endocrine organs • hypothalamus - important releasing factors that control pituitary gland |
|
|
Types of hormonal communication
|
• Autocrine - hormone is released and acts on receptors located on same cell
• Paracrine - hormone acts on neighboring cells • Endocrine - hormone travels through the bloodstream and acts on distant tissues with appropriate receptors Most, if not all, hormones have at least 2 of these actions, if not all 3. |
|
|
Control of Hormone Secretion
|
• negative feedback: most common: hormone directly or indirectly inhibits its further secretion
• positive feedback: uncommon: indirectly stimulates further secretion • releasing/inhibiting factors: common: hormones or biological effectors (glucose or Ca2+) • cyclical variations: common: e.g. diurnal cycle, sleep, seasons, etc |
|
|
thyroid disorders and feedback examples
|
• lack of neg feedback: TSH in a baby with no thyroid gland is extremely high (primary hypothyroidism)
• secondary hypothyroidism: not enough TSH to stimulate thyroid gland (e.g. malfunctioning pituitary or hypothalamus b/c of brain tumor) • Graves disease: body making antibodies binding to TSH receptor to tell it to make thyroid hormone like crazy -> hyperthyroid -> T4 very high, TSH will be low (neg feedback) |
|
|
Receptors
|
• hormone specificity is determined by receptors
• receptors are large proteins on cell membrane that determine responsiveness •location of receptors: cell surface, cytoplasm/nucleus |
|
|
Free vs Bound Hormones
|
• Bound hormone provides reservoir and extends the half life for stable hormone action
• Free hormone is active - available to bind to receptors and function in feedback regulation • binding proteins affect Total (bound) but not Free (unbound) hormone concentration • Equilibrium between bound: free hormone • albumin is non-specific binding protein, but other proteins are specific |
|
|
Free vs bound hormone as it relates to thyroid hormones
|
• 99% of thyroid hormone is carried by thyroid binding globulin (thyroid hormone itself is insoluble so needs to be carried), only free thyroid hormone can go into cell
•UNBOUND hormone is what controls/is controlled by pituitary gland: the FREE T4 has biologic action: e.g. if you have a mutation in thyroid binding globulin and your free T4 are normal, then your TSH is normal because pituitary gland is controlled by free T4 levels •If you are on birth control that stimulates you to make more thyroid binding globulin -> bind more free T4 -> pituitary senses low free T4 and makes more T4 to bring back to normal -> now total T4 is higher because you have more T4 bound to globulin, but same levels of free T4 because of feedback |
|
|
Growth hormone
|
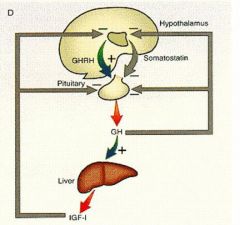
Hypothalamus releases GHRH -> pituitary secretes GH -> GH circulates and stimulates IGF-1 in liver and bone (muscle and protein synthesis). Then, IGF-1 feeds back and inhibits both hypothalamus and pituitary gland. Somatostatin secreted by hypothalamus inhibits pituitary gland from secreting GH.
|
|
|
Actions of Growth hormone
Bone |
• metabolism: increased osteoclast differentiation and activity, increased osteoblast activity, increase in bone mass by endochondral bone formation
• linear growth: increased epiphyseal growth, clonal expansion of osteoblasts |
|
|
Actions of Growth hormone
Muscle |
• increased amino acid transport
• increased nitrogen retention • increased metabolically active tissue and increased energy expenditure |
|
|
Actions of Growth hormone
Adipose tissue |
• acute insulin-like effects followed by increased lipolysis, inhibition of lipoprotein lipase, stimulated hormone sensitive lipase (HSL), decreased glucose transport, decreased lipogenesis
|
|
|
LH/FSH
|
LH/FSH normally very low in childhood. in puberty, hypothalamus releases GnRH -> stimulates pituitary to secrete LH and FSH, which stimulate ovaries to release estradiol/progesterone/ovulation, and stimulate testes to secrete testosterone, inhibin, spermatogenesis. Then, estrogen & testosterone neg feedback to hypothalamus & pituitary gland
|
|
|
Adrenocorticotropin hormone (ACTH)
|
Hypothalamus releases CRH -> pituitary releases ACTH -> adrenal glands secrete cortisol, which negatively feeds back to both hypothalamus and pituitary
|
|
|
adrenal gland
|
salt, sugar sex
• glomerulosa: aldosterone - regulates salt uptake in kidney • fasciculata - secretes cortisol • reticularis - makes androgens at time of puberty (testosterone-like, gives girls pubic hair and axillary hair) • medulla - sympathetic ganglion that invades outer cortex makes epinephrine for fight/flight response |
|
|
Cortisol
|
crucial for glucose metabolism, protein/fat metabolism, blood pressure
• If adrenal insufficiency (lack of cortisol) -> ACTH build up is high (primary adrenal insufficiency - hypertension, shock) • If stress -> increased CRH from hypothalamus -> ACTH increases -> circulating cortisol can incr from 10 mg/dL to 100 mg/dL |
|
|
Thyroid stimulating hormone
|
Hypothalamus releases TRH -> pituitary releases TSH -> thyroid gland makes T3 and T4. Like in GH pathway, somatostatin secreted by hypothalamus inhibits pituitary.
• thyroid gland follicles consist of lumen containing thyroglobulin (TG) protein which was formed by thyrocytes. TG can make T3 and T4 by using thyroid peroxidase to attach iodine (originally transported into lumen from blood stream via Na+/I- symporter) to tyrosine. Then, the T3/T4 goes back into thyrocyte colloid and gets pinched off as it gets dumped into blood stream. T4>T3, secreted into blood, carried by TBG. |
|
|
Insulin secretion pathway
|
Eat food -> glucose transporter transfers glucose into cell -> G6P undergoes glycolysis, krebs cycle, and eventually makes ATP -> ATP build up inhibits ATP-sensitive K+ channels -> cell is depolarized -> Ca2+ rushes in, then stimulates release of insulin
|
|
|
Food, calcium and PTH
|
• Eat food -> high levels of Ca2+ in blood -> thyroid gland parafollicular cells release calcitonin -> calcitonin inhibits osteoclasts, decreasing blood Ca2+ level -> low Ca2+ levels stimulates parathyroid gland chief cells to release PTH -> PTH promotes osteoclasts to resorb Ca2+ from bone, slows down loss of Ca2+ in urine (via kidney), stimulates kidneys to release calcitrol which incr Ca2+ binding protein in gut which leads to more uptake of Ca2+ from food in gut.
|
|
|
Renin-angiotensin-aldosterone cascade
|
blood volume low -> macula densa in distal tubule senses drop in pressure & [NaCl] -> juxtaglomerular cells on afferent arteriole secrete renin into circulation -> renin converts angiotensinogen released by liver to angiotensin I -> ACE converts angiotensin 1 to angiotensin 2 -> angiotensin 2 causes vasoconstriction and also stimulates secretion of hormone aldosterone from adrenal cortex -> aldosterone acts on distal tubule of kidney to increase reabsorption of Na+ (and therefore water) into blood -> increase bp
|
|
|
Hormones regulate four domains
|
• Reproduction: LH, FSH, estrogen, testosterone, prolactin
• Internal environment: vasopressin, parathyroid hormone, aldosterone • Growth and development: Growth hormone, thyroid hormone, estrogen, DHEAS, testosterone • Energy production, metabolism: insulin, glucagon, cortisol, thyroid hormone |
|
|
Types of Endocrine Disorders
|
• Deficiency - usually decreased production (primary vs secondary, congenital vs acquired)
• Excess - usually overproduction (e.g. tumors or autoimmune disease) • Resistance - receptor/second messenger defects: congenital or genetic • Cancer: inherited or spontaneous |
|
|
Diabetes
|
• Type II diabetes - insulin resistance - receptors are not recognizing insulin, so B cells of pancreas have to produce more and more insulin to keep blood sugars normal -> eventually B cells “conk out” and can’t make enough insulin -> blood sugar rises and you get type 2 diabetes
• Type 1 diabetes is Islet B cell destruction, whereas Type 2 is insulin resistance with B cell secretory defect |
|
|
Properties of steroids
|
• Steroids are a class of terpenes, which have carbon skeletons made up of four fused rings in 6 carbon-6 carbon-6 carbon-5 carbon formation:
• Steroids are hydrophobic, so they are said to be “lipid-like” |
|
|
Animal Steroids include
|
• Sterol (cholesterol)
• Bile acids • Steroid hormones Most steroid hormones are derived from cholesterol |
|
|
Four parent ring structures are precursors for important products
|
• Cholestane (C-27) makes cholesterol
• Pregnane (C-21) makes progesterone • Androstane (C-19) makes androgens • Estrane (C-18) makes estrogens |
|
|
Stereochemistry
|
the spatial arrangement of atoms within molecules
• Important for reactivity and specificity E.g. 5α-dihydrotestosterone is an active androgen, but 5β-dihydrotestosterone has no biological activity due to its configuration. |
|
|
Cholesterol biosynthesis
Location |
• Virtually all tissues, but particularly high in liver, adrenal cortex, and gonads
• Synthesis occurs in the cytoplasm (cytosol and ER) [N.B. this is a similar pathway as keto-acid synthesis. Keto-acid synthesis occurs in mitochondria, whereas cholesterol synthesis occurs in the cytosol.] |
|
|
Cholesterol biosynthesis
substrates |
• NADPH is required as a reducing equivalent
• Coenzyme A (CoA) and ATP provide energy • ATP also donates phosphate groups |
|
|
Cholesterol biosynthesis
important enzymes |
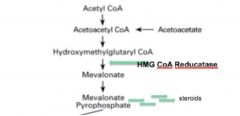
• HMG CoA Reductase catalyzes the formation of mevalonate, which is the key rate-limiting step in cholesterol synthesis.
• This enzyme is a common drug target • After the synthesis of mevalonate, the cholesterol synthesis pathway is constitutively active |
|
|
Cholesterol is NOT an energy source
|
(cannot be degraded into CO2 and water.) It is instead metabolized in the form of bile salts and bile acids and excreted.
|
|
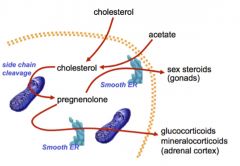
STEROID HORMONE BIOSYNTHESIS FROM CHOLESTEROL
Localization |
•Cholesterol is either derived from plasma or synthesized in the smooth ER and cytoplasm
•Cholesterol is transported to the mitochondria for side-chain cleavage, forming pregnenolone. •Pregnenolone can then return to the smooth ER for further modification in certain tissues: • In the gonads, pregnenolone is converted to androgens, estrogens, and progestins • In the adrenal cortex, substrates downstream from pregnenolone are converted to glucocorticoids and mineralocorticoids. |
|
|
Cytochrome p450 enzymes
|
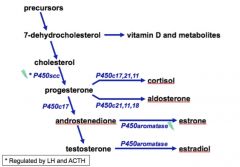
• P450 side chain cleavage enzyme (P450scc)
• Catalyzes the conversion of cholesterol to progesterone • Regulated by LH in the gonads • Also regulated by ACTH in the adrenal cortex |
|
|
StAR protein
|
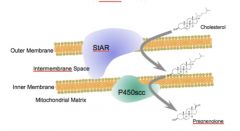
•Acutely regulates steroid hormone synthesis by aiding P450scc in side-chain cleavage.
•Specifically, this protein transports the hydrophobic cholesterol molecule across both the outer and inner mitochondrial membrane, where P450scc can convert it to pregnenolone (and progesterone downstream.) •The StAR and P450scc proteins are contiguous with one another across the mitochondrial membranes. |
|
|
Defects in steroid hormone biosynthesis
|
• Lipoidal Congenital Adrenal Hyperplasia
• Smith-Lemli-Opitz Syndrome • Congenital Adrenal Hyperplasia (CAH) • 5α-Reductase deficiency |
|
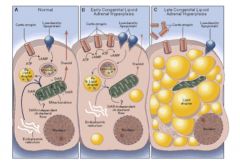
Lipoidal Congenital Adrenal Hyperplasia
|
• A genetic loss of ALL steroidogenesis from cholesterol due to a defect in either StAR protein or P450scc enzyme.
• Fatal if not treated early in infancy • All affected individuals are phenotypic females because they make no androgens during development • Associated with severe salt-losing due to absence of mineralocorticoids • This can now be diagnosed prenatally on a newborn screen • In the adrenal gland, you get an increase of ACTH because of a lack of negative feedback by steroid hormones. Secondary fat (=lipodal) accumulation occurs, causing hypertrophy of the adrenal gland (=adrenal hyperplasia.) |
|
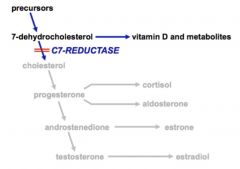
Smith-Lemli-Opitz Syndrome
|
An autosomal recessive disorder caused by a deficiency in C7-Reductase enzyme
o Cannot synthesize androgens, mineralocorticoids, or glucocorticoids o Results in low cholesterol and accumulation of 7-dehydrocholesterol o Simply supplementing cholesterol does not ameliorate the condition o Mental retardation o Ambiguous genitalia in males (due to lack of testosterone production) o Salt and sugar-losing due to lack of mineralo- and glucocorticoids |
|
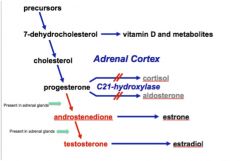
Congenital Adrenal Hyperplasia (CAH)
|
• Most commonly due to a deficiency in C21-hydroxylase, an enzyme that normally converts progesterone to cortisol or aldosterone in the adrenal cortex.
o Lack of negative feedback by cortisol and aldosterone causes increased release of CRH from the pituitary, ultimately leading to adrenal hyperplasia o Progesterone in the adrenal cortex that cannot be converted to cortisol or aldosterone is shunted into the alternate androgenic pathway. o Androstenedione and testosterone levels will be high because the enzymes to make these from progesterone are present in the adrenal cortex. Estrone and estradiol will be unaffected, as the enzymes that make these hormones are not in the adrenal cortex o Cortisol and aldosterone levels will be low |
|
|
Congenital Adrenal Hyperplasia (CAH)
clinical presentation |
o Salt-washing crisis in the first weeks of life, can be fatal if left untreated
o Girls become viralized due to the overproduction of androgens and present with ambiguous genitalia o Boys are phenotypically normal, which is dangerous as the CAH can be unrecognized and the infant is at risk of dying from shock early in life (from lack of gluco- and mineralocorticoids) o Treatment is done by replacing gluco- and mineralocorticoids. |
|
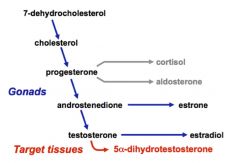
5α-Reductase deficiency
|
• An autosomal recessive disorder expressed in 46,XY males causing a mutation in 5α-reductase-2, which normally converts testosterone to 5α-dihydrotestosterone (a more potent virilizing hormone in males.)
o Males born undervirilized, but become virilized at puberty o Usually have ambiguous genitalia, with blind vaginal pouch o Wolffian derivatives are normal and Mullerian derivatives are absent o Normal testes are present |
|
|
MECHANISM OF STEROID HORMONE ACTION
receptors |
• Steroids are hydrophobic so they can cross the plasma membrane. They usually activate receptors in the cell that can directly act as transcription factors.
•Different families of steroid hormone receptors are specific for certain ligands. |
|
|
MECHANISM OF STEROID HORMONE ACTION
mechanism |
•A steroid hormone binds to its receptor in the cell, causing phosphorylation, conformational change, and release of heat-shock protein (hsp) from receptor.
•Receptor dimerizes with another activated hormone receptor •Steroid response element (SRC) protein coactivates the dimerized complex •This complex goes on to very specifically bind DNA via its “zinc finger” domain and activate transcription |
|
|
Steroid and Thyroid hormone receptors are considered to be part of the same superfamily meaning..
|
• DNA binding domains would be highly conserved
• Hormone binding domains would NOT be conserved--they would be specific to their hormone. |
|
|
Complete Androgen Insensitivity
|
X-linked recessive disorder in 46,XY individuals caused by a mutation in the androgen receptor (AR) gene
Disorder of sexual differentiation: patients have: o Breast development and female hiatus at puberty o Primary amenorrhea o Scant or absent pubic and axillary hair o Female genitalia with blind vaginal pouch o Wolffian derivatives usually absent, Mullerian absent or vestigial o Testes present |
|
|
Anabolic steroids
|
• Anabolic steroids are naturally or synthetically occurring androgens
• Testosterone levels in most men almost totally saturate the androgen receptors in target tissues, so the effects of anabolic steroids probably use another mechanism |
|
|
How Anabolic steroids might work:
|
Fully saturating the few androgen receptors that are unbound (can lead to a paltry 2-5% increase in muscle mass over controls)
•Blocking the effect of glucocorticoids on muscle, which normally stimulate breakdown of protein |
|
|
Signal transduction definition
|
• How a cell uses extracellular changes to get internal, cellular functional changes
• Hormones can either bind intracellularly directly or bind high affinity cell surface receptors • Once a hormone binds the cell surface receptor, a second messenger has to assist; the first messenger is the hormone itself ex: epinephrine (hormone) activating phosphorylase which activates glycogenolysis in the cell |
|
|
cAMP
generation and degradation |
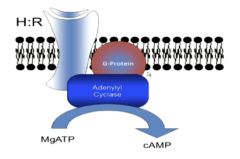
•cAMP generation (highly favored): comes from Mg(2+)-ATP via adenylyl cyclase (it cyclizes it and links an OH-methyl group on the 5’ to the 3’ phosphate
•cAMP is degraded by cAMP phosphodiesterase into AMP |
|
|
Regulation by epinephrine (hormone)-How an external molecule affects cAMP production
|
•Epinephrine binds its hormone receptor on the surface, ß-adrenergic receptor to form an H:R complex on the cell surface
• Guanosine Triphosphate (GTP) is bound • GTP has two simultaneous functions: 1) it allows activation of adenylyl cyclase to form our second messenger, cAMP and gets converted to GDP and 2) causes dissociation of the hormone and receptor |
|
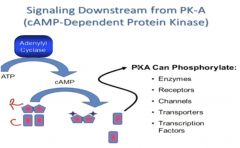
Signaling by cAMP
|
•cAMP mediated by cAMP dependent protein kinase (Protein Kinase A or PKA), a serine/threonine kinase
• PKA can control transcription, enzymes, receptors, ion channelfs (phosphorylation by PKA leads to DNA binding) •Transcriptional control: many promoters have cAMP response elements, cAMP response element binding protein (CREB) activated |
|
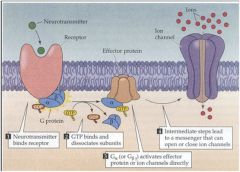
G Protein Signal Transduction
• Role of G-Protein |
• bind GTP
• activate adenylyl cyclase (cAMP generation) • cause dissociation of epinephrine from ß-adrenergic receptor • hydrolyze to GDP to inactivate themselves • Binding to GTP is the ON signal, Hydrolysis to GDP is the OFF signal |
|
|
Gprotein subunits: The Heterotrimeric G-Protein
|
• G-protein has 3 subunits: 𝞪,𝞫,𝞬
• Two types of alpha: Gs𝞪 (stimulates adenylyl cyclase) and Gi𝞪 (inhibits adenylyl cyclase) • The alpha subunit converts GTP to GDP and can separate from the complex to bind other effectors |
|
|
G-protein Heterogeneity
|
due to alternative splicing and multiple genes, many different types of g-proteins.
relay signals for over 1000 receptors, many different families and combinations of subunits |
|
|
Rhodopsin/Transducin System
|
Light reception
•Analagous to receptor/g-protein/cyclase system • Rhodopsin=receptor, Transducin=G-protein, Phophodiesterase=effector like cyclase (changes cGMP to 5’GMP) • Lower cGMP levels→closing sodium channels→hyperpolarization of visual cortex |
|
|
Olfaction
|
G(olf) is a special G-protein. The olfactant binds to a chemoreceptor, G(olf) activates adenylyl cyclase (ATP→cAMP), which leads to a sodium channel being opened for transmission of a signal down an axon
|
|
|
Cholera
|
• secretory diarrhea; cholera toxin is local to gut
• toxin=enzyme, causes ADP-Ribosylation on an Arg Residue of Gs𝞪 • No GTPase; Adenylyl Cyclase stimulated |
|
|
Pertussis
|
• whooping cough, hypoglycemia; pertussis toxin is systemic
• toxin is also an enzyme that causes ADP-Ribosylation on Gi𝞪 • No inhibition of Adenylyl Cyclase; no ability to exchange GTP for GDP |
|
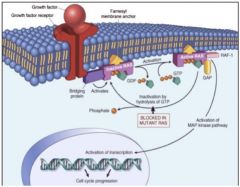
Cancer
|
• Via distant relatives, “small G-proteins” Ras
• Ras is a signaling protein that has characteristics of a G-protein; Ras is activated by a growth factor binding to a growth factor receptor, and once activated with GTP, it upregulates growth genes •Cancer is uncontrolled growth; the GTPase activity is lost, it stays on all the time, and mutant Ras allows growth to continue |
|
|
Pseudohypoparathyroidism
|
truncated Gs𝞪 protein; elevated PTH made him short and had seizures and obesity
|
|
|
McCune Albright Syndrome
|
autonomous endocrine gland due to activating Gs𝞪 defect
|
|
|
Signal Transduction Definition
|
• Signal transduction is the system by which an extracellular hormone induces intracellular hormones
• Relies on different components: the First messenger (the hormone itself) the Second Messenger (cAMP), Adenylyl Cyclase (makes cAMP from Mg-ATP), and the G-Protein (binds GTP and activates adenylyl cyclase) |
|
|
Second Messengers
|
•free ATP is cyclized by adenylyl cyclase into cAMP
•PKA binds cAMP and regulates it to change transcription, enzymes, channels, and receptors • PKA made of 2R-2C; 2R binds cAMP, 2C=transfers phosphate from ATP • This is how cAMP controls the cell! • PKA is a serine/threonine kinase |
|
|
Signal Transducers (G-protein)
|
•The critical middle-man: G-protein binds GTP to turn the system “on”; hydrolyzes GTP to GDP to turn “off”
•G-protein made of 𝞪,𝞫,𝞬 subunits; 𝞫,𝞬 stay together next to the cell-surface receptor, 𝞪 controls adenylyl cyclase • Two types of alpha: Gs𝞪 (stimulates adenylyl cyclase) and Gi𝞪 (inhibits adenylyl cyclase) |
|
|
Light Reception/Olfaction
|
•Both have analogous systems
•Light: Rhodopsin=receptor, Transducin=G-protein, Phophodiesterase=effector like cyclase (changes cGMP to 5’GMP) •Olfaction: G(olf) is a special G-protein; cAMP leads to a sodium channel being opened |
|
|
Infectious Disease, Cancer, and other disorders
|
•Cholera=ADP ribosylation on Gs𝞪 to turn of GTPase and keep Adenylyl Cyclase going
•Pertussis=ADP ribosylation on Gi𝞪 to not putting in GDP and keeps Adenylyl Cyclase going •Cancer=distant “cousins” of G-proteins, small G-protein Ras mutated. GTPase activity lost due to mutant Ras, and growth allowed to go on without stopping |
|
|
NON-RECEPTOR TYROSINE KINASE: SRC
a. Phosphorylation |
• Reversible phosphorylation occurs on serine, threonine, and tyrosine, but only a small proportion is on tyrosine (0.1-2%)
• Tyrosine phosphorylation was shown to be important from cancer-causing genes that were transmitted by a virus |
|
|
NON-RECEPTOR TYROSINE KINASE: SRC
b. The Virus Connection |
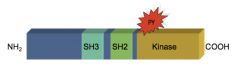
• Rous sarcoma virus, a retrovirus, caused “cell transformation” when inserted into a cell
• cell transformation=cancer: loss of contact inhibition, don’t need growth factors, immortal, increase glycolysis and glucose transport, no anchorage dependence • One protein product caused cell transformation: v-Src, a kinase that phosphorylated a tyrosine residue •The protein kinase v-Src phosphorylated itself as a substrate |
|
|
Normal, Human Analog to v-Src
|
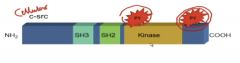
• c-src is a normal, human homologue to v-src
• Although expressed at 15X normal does not cause cell transformation • Due to autophophorylation site on Tyr527 (see image above, double circle); acts as an inhibitory site • This site is missing in v-src, and it is on all the time (no inhibition) |
|
|
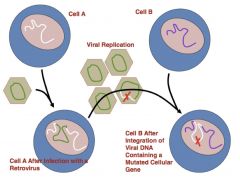
c-src was mutated in viral replication to create v-src (loss of carboxy-end inhibition site)
|
|
|
|
most human cancers are just spontaneous mutations of proto-oncogenes (normal human gene) into oncogenes (causes cancer):
|
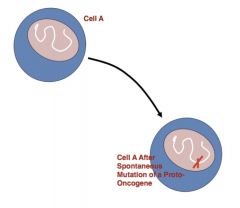
|
|
|
SIGNAL TRANSDUCTION FROM Epidermal Growth Factor (receptor Tyrosine Kinase)
|
• A diffusible growth factor that comes from salivary glands to open mice eyes (closed at birth)
• The EGF receptor has the prototypical tyrosine kinase structure • EGF has many of same effects as Src • growth stimulation, increased nutrient transport, cell differentiation, activation of glycolysis |
|
|
EGF receptor structure
|
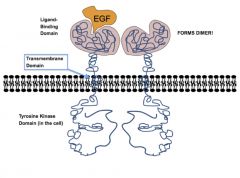
3 main components (similar to G-protein!):
1) Extracellular ligand binding domain 2) transmembrane domain 3) tyrosine kinase domain (with phosphorylation sites, substrate binding regions) |
|
|
EGF receptor function
|
• Tyrosine domain is required for biological activity of the receptor: studies done on tyrosine-domain ATP binding site mutation leading to loss of function
•When a ligand binds to receptor (e.g. EGF binding to EGF-receptor), serial conformational changes in whole molecule (Transmembrane→Tyrosine Kinase) •Dimer forms with other EGF receptor molecule, and the two tyrosine kinases “trans-phosphorylate” each other on tyrosine |
|
|
HER Family
|
Related Tyrosine Kinase
• HER1=EGF receptor (HER4, HER3 also bind ligands) • HER2 doesn’t bind ligands • HER3 has no tyrosine kinase, but can still dimerize • One HER that can bind ligands dimerizes with HER2, and they phosphorylate each other • Signals go to nucleus to increase transcription (VEGF=growth factor for increased vessels) |
|
|
“Trastuzumab”
|
a drug, binds HER2 stopping its function (survival lengthened for breast cancer patients from 23 to 25 months)
|
|
|
EGF Receptor and Erb-B
|
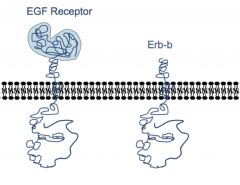
• erb-b is a truncated EGF receptor
• Trunctation=non-regulated tyrosine kinase • Avian erythroblastosis virus and breast cancers use the erb-b family to cause cancer |
|
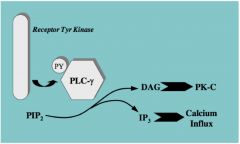
RECEPTORTYROSINE KINASE: CELL SIGNALING DOWNSTREAM
Mechanism 1: Direct Activation |
•Phospholipase C-𝞬 (PLC𝞬) activated by phosphorylation on a tyrosine residue
•PLC𝞬 hydrolyzes PIP2 into DAG and IP3 •DAG activates Protein Kinase-C (analogous to cAMP activating cAMP-dependent protein kinase) •The conversion of PIP2 to IP3 raises cell calcium levels. • The reason this is such a breakthrough is because it’s making a connection between activation of a receptor kinase and something happening in the cell that can mediate an effect. General Principle: A receptor phosphorylates an enzyme on tyrosine (activates it); the enzyme results in transduction of a signal |
|
|
RECEPTORTYROSINE KINASE: CELL SIGNALING DOWNSTREAM
Mechanism 2 |
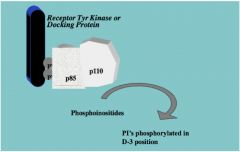
• p110 is an enzyme PI3 Kinase
• Catalyzes phosphoinositides in the cell getting phosphorylated in the 3rd position • Difference between this mechanism and the previous mechanism is that PI3 Kinase is not itself phosphorylated, its regulatory subunit, p85, is phosphorylated to activate p110 |
|
|
RECEPTORTYROSINE KINASE: CELL SIGNALING DOWNSTREAM
Mechanism 3: Ras Proteins |
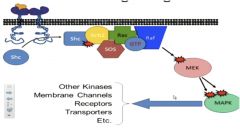
• Growth factor receptor activates a bridging protein that promotes nucleotide exchange
• Ras is activated • Growth factor binds →autophosphorylation on tyrosine • Tyrosine phosphorylation→Proteins aggregate |
|
|
Shared domains for different mechanisms• SH2 interact with tyrosine phosphorylated proteins
• SH3 interact with cytoskeletal protein |
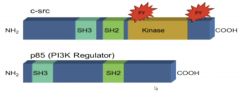
|
|
|
RECEPTORTYROSINE KINASE: CELL SIGNALING DOWNSTREAM
Mechanism 4: JAK/STAT System |
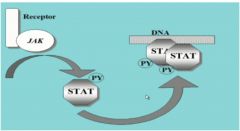
•Receptor bound to kinase, JAK (“Just Another Kinase”)
•phosphorylates STAT, acts rapidly to induce gene expression •Almost like a short circuit with two steps |
|
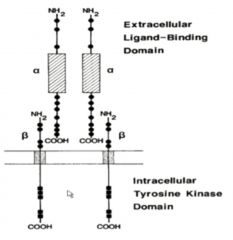
INSULIN RECEPTOR
Structure |
• Already dimerized
• Alpha extracellular, Beta transmembrane, internal tyrosine kinase • Tyrosine Kinase substrates are IRS-1, IRS-2… • Metabolic effects of Insulin involve dephosphorylation via activation of protein phosphatases |
|
|
INSULIN RECEPTOR
signaling |
• Promotes uptake of glucose by skeletal muscle
• PI-3 Kinase bound to IRS-1 • PI-3 Kinase signaling increases number of available glucose transporter by allowing internal membranes to surface • Only if GLUT4 is the glucose transporter will it be sensitive to insulin |
|
|
summary
• Non-Receptor Tyrosine Kinase: Src |
• Tyrosine kinase importance in causing cell transformation discovered from Rous Sarcoma virus causing cell transformation (cancer)
• Isolated gene, v-Src a tyrosine kinase • Human analogue, c-Src, had a phosphorylation site on carboxy end that inhibited |
|
|
summary
• EGF-Receptor & Tyrosine Kinase |
• Growth Factor Receptors connected to a tyrosine kinase to have cellular effects
• Discovered from studying growth of skin cells on mice eyes to open them; due to Epithelial Growth Factor • 3 main components (similar to G-protein!): 1) Extracellular ligand binding domain 2) transmembrane domain 3) tyrosine kinase domain (with phosphorylation sites, substrate binding regions) oTyrosine domain is required for biological activity of the receptor • Dimerization allows transphosphorylation • HER family uses similar mechanism, where different HER molecules can/cannot bind ligand, but dimerize • erb b= truncated EGF-receptor (breast cancer, avian erythroblastosis virus) |
|
|
summary
• Receptor Tyrosine Kinase: Cell Signaling Downstream • Mechanism 1 |
Phospholipase C-𝞬 (PLC𝞬) activated by phosphorylation on a tyrosine residue
o PLC𝞬 hydrolyzes PIP2 into DAG and IP3 o DAG activates Protein Kinase-C (analogous to cAMP activating cAMP-dependent protein kinase) |
|
|
summary
• Receptor Tyrosine Kinase: Cell Signaling Downstream • Mechanism 2 |
p85 phosphorylated, activates p110 (Pi3 Kinase)
o Catalyzes phosphoinositides in the cell getting phosphorylated in the 3rd position |
|
|
summary
• Receptor Tyrosine Kinase: Cell Signaling Downstream • Mechanism 3 |
Growth Factor binds, autophosphorylation on tyrosine→Proteins aggregate (effects Ras)
|
|
|
summary
• Receptor Tyrosine Kinase: Cell Signaling Downstream • Mechanism 4 |
Receptor bound to kinase, JAK (“Just Another Kinase”), phosphorylates STAT, acts rapidly to induce gene expression
|
|
|
summary
•Receptor Tyrosine Kinase: Cell Signaling Downstream Shared domains |
SH2 interact with tyrosine phosphorylated proteins, SH3 interact with cytoskeletal proteins
|
|
|
Insulin Receptor
|
• dimerized, alpha extracellular, beta transmembrane, internal tyrosine kinase
• tyrosine kinase acts on IRS-1, IRS-2 • PI-3 Kinase bound to IRS-1, increases available number of glucose transporters |
|
|
Basic cell cycle regulation ideas
|
a. Check point after G1 before entering S
b. doubling the amount in the cell; get ribosomal biogenesis in order to double amount of ribosomes; ribosome levels are what cell detects to see if it’s doubled in size (sufficient growth) c. Limited by: nutrients, “crowdedness” d. Goes into G0 if doesn’t pass the check point e. CC: cancer cells can bypass this checkpoint f. Senescence: permanent growth arrest; often happens after differentiation; eg white adipose, muscle, brain cells, cardiac cells-Aka terminal differentiation g. Cells can also enter apoptosis |
|
|
Cell cycle components
|
3 families:
cyclins, cyclin dependent kinases (CDK), CDK inhibitors |
|
|
Cyclins
|
activating regulatory subunits of cyclin/CDK complexes
•They upregulate and downregulate during phases of cell cycle; they’re specific for certain phases and interact with CDK and either activate or inhibit it |
|
|
Cyclin Dependent Kinases (CDK)
|
signaling kinases of cell cycle; mostly affect serine/threonine
|
|
|
CDK inhibitors (CKIs)
|
inhibITORY subunits of cyclin/CDK complexes
• Most important for apoptosis and cell differentiation • Bind to cyclin/CDK complexes to inhibit CDK catalytic activity • 2 major classes: K.IP and INK There are many members of each class, and much overlap btwn classes and members |
|
|
CAK: CDK activating kinase
|
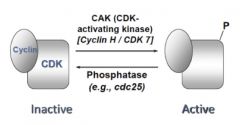
Sometimes, CDK/cyclin complexes must be activated by kinases to actually become active
|
|
|
EF2/Rb
|
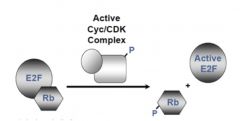
inactive when complexed together
When active cyclin/CDK complex (so it’s been phosphorylated) phosphorylateS Rb, Rb-P leaves and allows EF2 to transcribe proteins |
|
|
general cell cycle
|
|
|
|
Cell cycle dysregulation in cancer
|
-Overexpression of cyclins
-Mutation inactivating Rb -Sequestration of Rb -CKI mutations |
|
|
Overexpression of cyclins
|
tend to affect G1 --> S restriction point
• Breast cancer • Leukemia • testicular cancer |
|
|
Mutation inactivating Rb
|
(so EF2 runs rampant)
• Retinoblasta: malignancy of retinal; loss of red reflex; congenital; screened for at birth • Small cell lung carcinomas • Osteosarcomas |
|
|
Sequestration of Rb
|
by E7 protein of HPV
• Completely different from what Ras Sarcoma virus do, which is affect tyrosine kinase receptor • Cervical cancer |
|
|
CKI mutations
|
• Prostate cancers, etc
|
|
|
Cell growth dysregulation in cancer
|
Cancer cells often very small; don’t wait until they’ve doubled in size before dividing
a. Altered regulation of ribosome synthesis (remember ribosome levels are often how cell can tell if it’s doubled in size) •Some cancer drugs inhibit ribosome biogenesis b. Can augment nutrient transport so nutrient availability isn’t limiting for growth •Eg upregulating amino acid transporters |
|
|
Growth
|
Growth in length – long bone
Growth in somatic mass – body weight • Cell ‘growth’ (cell gets bigger; aka hypertrophy) • Cell proliferation (cells get more numerous) |
|
|
bone growth
|
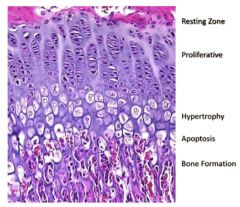
|
|
|
growth signals
|
Normally, hypothalamus releases GHRH to stimulate pituitary to release GH, which stim IGF-1 to cause epiphyseal plate development
• If you take out hypothalamus, lose the epiphyseal plate |
|
|
Replicative senescence
|
limit to number of times cells in bone resting zone can divide
• Timing of replicative senescence in this zone is variable btwn individuals |
|
|
what regulates replicative senescence
|
Estrogen regulates senescence in resting zone via Primary Estrogen Receptor
• This is why women stop growing when menarche starts (bc get estrogen) • While boys can still grow after puberty (hair and testes; androgen-dependent not estrogen dependent) |
|
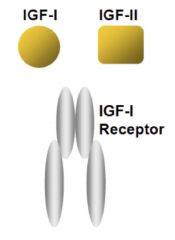
IGF-1 receptor
|
heterodimer; coded for by IGF-1 and IGF-2
• If you KO IGF-1, get a 60% size mouse • If you KO IGF-2, get a 60% size mouse • If you KO both, get a 30% size mouse • If you KO IGF-1 receptor, get 45% mouse • These mutations in IGF system cause decrease in cell number, not cell size |
|
|
fun facts about IGF
|
When people fast, their IGF-1 goes down
When they eat, the IGF-1 goes up, correlating with the amount of protein they ate This increase in protein in the diet is why people who move to the US grow taller, regardless of race Difference in sizes between dogs can be accounted for by IGF-1 alleles Obese boys tend to enter puberty later (so usually taller), and obese girls tend to begin breast development earlier (so usually shorter) |
|
|
Euchromatin
|
When cell is in interphase, chromatin is loose; allows active transcription
|
|
|
heterochromatin
|
condensed chromatin
|
|
|
23 chromosomes
|
• 22 autochromes
• 1 sex chromosome • We’re diploid so a pair of each chromosomes • Gametes are haploid (1N) • N = number of chromosomes • C = number of chromatids |
|
|
Mitosis check points and cell cycle steps
|
G1 most variable
If conditions not right after G1, can also go to G0 a. S: genetic material duplicates b. G2 preps for mitosis c. Mitosis: actual division and partition d. Checkpoints at G1-S and G2-M |
|
|
mitosis phases
|
Prophase
metaphase anaphase telophase cytokinesis |
|
|
Prophase
|
chromatin condenses to form well defined chromosome (made of 2 chromatids, a centromere), mtoc (centrosome complex; microtubule organizing center) splits into 2 and bcms polarized
|
|
|
Prometaphase
|
nuc envelope disrupted, kinetochores assemble, kinetochore, polar, astral microtubules
|
|
|
Metaphase
|
chromosomes align; do karyotype analysis at this stage; cells can be arrested with colchicine, a microtubule inhibitor
|
|
|
Anaphase
|
centromeres are pulled apart
|
|
|
Telophase
|
nuclear env reforms, chromosomes decondense, spindle fibers disappeares
|
|
|
Cytokinesis
|
Cleavage furrow
|
|
|
Meiosis
|
1 round DNA synth
2 rounds segreg and div |
|
|
Meiosis 1
|
homologous chromosomes segreg into daughter cells (2 daughter cells)
prophase I: crossing over happens here |
|
|
meiosis II
|
chromatids from each chromosome segregate to daughter cells (4 daughter cells)
cells become haploid |
|
|
N and C
|
mitosis: go from 2N, 2C --> (replicate DNA) 2N, 4C --> (post-cytokinesis) 2N, 2C
meiosis: go from 2N, 2C--> (replicate DNA) 2N, 4C à (post-meiosis I) 2N, 2C --> (post-meiosis II)1N, 1C |
|
|
Prophase 1 subphases
|
Leptotene
Zygotene Pachtytene Diplotene Diakineses (dictyotene in oogenesis) |
|
|
leptotene
|
chromosomes already replicated during S phase; condensation starts
|
|
|
zygotene
|
synapsis; they pair; held together by protein structure called synaptonemal complex
|
|
|
pachtytene
|
meiotic crossing over takes place
|
|
|
diplotene
|
synaptonemal complex disappears; homologs held together with chiasmata
|
|
|
diakinesis
|
shortening and thickening of paired chromosome; formation of spindle fibers
|
|
|
dictyotene
|
which is prolonged resting phase after diplotene in oogenesis
|
|
|
spermatogenesis
|
spermatogonium: sperm stem cell;
-forms primary spermatocytes (2N, 4C; duplicated DNA) à secondary spermatocytes (1N, 2C) à spermatid and then spermatozoan (1N, 1C) |
|
|
oogenesis
|
division of cytoplasm is unequal; so instead of 1 diploid --> 4 haploid gametes, 3 of those haploids are non-viable bc don’t have enough cytoplasm
oogonium (2N, 2C) --> primary oocyte (2N, 4C; arrested in prophase I until puberty; re-starts right before ovulation) --> secondary oocyte (1N, 2C; arrested in metaphase II until fertilization) --> ovum haploid |
|
|
chromosomal disorders
|
Subset of genetic disorders
• Others include single gene inheritance, multifactorial, and mitochondrial |
|
|
homologous chromosomes
|
members of pair of chromosomes carrying matching genetic information
• Same genes in same sequence, but at any specific locus, may have either identical or slightly different forms of same gene (alleles) |
|
|
Karyotype
|
arrangement of chromosomes by size/morphology
• Chromosomes best studied while in metaphase • G-banding (characteristic patterns) created by trypsin (protease) and Giemsa (DNA staining chemical) • Arranged in pairs from largest (1) → smallest (22) • Short arm = p; long arm = q |
|
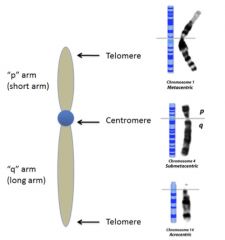
p and q
|
o Metacentric: equal lengths
o Submetacentric: clear short + long arms ( → pq designation) o Acrocentric: little to no short arm with prominent long arm |
|
|
International System for Human Cytogenetic Nomenclature (ISCN)
|
Karyotype components: # chromosomes, sex, change 1, change 2, etc. (if applicable)
p = short arm q = long arm + = additional chromosome - = missing chromosome t = translocation del = deletion dup = duplication inv = inversion der = derivative = structurally-arranged chromosome |
|
|
X-chromosome inactivation
|
males have 1 X-chromosome, while females have 2
• Only 1 X-chromosome in each cell within female body is actively transcribed (used) → Barr Body: inactive (condensed) X-chromosome # X chromosomes (NOT # Y) dictate # Barr Bodies (in equation form, X-1) • Ex: XXX has 2 BBs, XXY has 1 BB, XX has 1 BB |
|
|
Incontinentia Pigmenti (IP)
|
mutation on IKBKG gene (X-linked dominant)
Females: Some cells express genes while other don’t => disperse pigmentation Males: mostly lethal (usually → miscarriage) Characteristic skin lesions • Birth → 4m: blistering • ~4 → 6m: wart-like rash • 6m → adult: swirling hyperpigmentation • → Adult: linear hypopigmentation Alopecia (hair loss) Hypodontia (↓ # teeth) Dystrophic nails (misshapen) Childhood retinal detachment risk Intelligence: mostly normal, some have intellectual disability/seizures |
|
|
non-disjunction
|
o “Not coming apart”: failure of chromosome pairs to separate properly during cell division → homologous chromosomes in same daughter cell
o Increased risk with advanced maternal age |
|
|
meiosis 1 and 2 non disjunctions
|
meiosis 1 :no normal gametes
meiosis 2: possible normal gametes |
|
|
Consequences of trisomies/monosomies
|
• Trisomies 21 (Down syndrome), 18, 13: Compatible with live birth (other autosomes are not)
• XXY: Klinefelter syndrome • 45, X: Turner syndrome |
|
|
trisomy rescue
|
loss of 1 chromosome n trisomy
o Possible → uniparetal disomy: normal # chromosomes, but from 1 parent o Possible → autosomal recessive disorder (to be continued) |
|
|
Triploidy
|
69 chromosomes (3 sets, not compatible with life)
• Frequent in missed abortion material |
|
|
Mitotic non-disjunction
|
(in zygote, NOT egg/sperm!) → clustering of affected cells → mosaicism (error in Mitosis)
|
|
|
Trisomy 21: Down Syndrome
|
Incidence: 1 in 650
Karyotype: •95% sporadic (extra chromosome) - nonfamilial •3-4% unbalanced translocation (usually 14, 21) • 75% de novo • 25% familial •1-2% mosaic Important to distinguish to give parents accurate recurrence risk |
|
|
Down syndrome characteristics
|
Facial appearance:
•Brachycephaly (“flat head”, or shorted ventral → dorsal) •Palpebral fissures (of eyes) slanted upward •Prominent epicanthal folds (of eyes, nearer nose) •Flat nasal bridge •Small ears •Flat facial profile •↓ mouth muscle tone → “open mouth” look Hypotonic (“floppy”) Nuchal skin fold (back of neck) Small size Single transverse palmar creases (however, found in 5% general population - NOT DIAGNOSTIC) Sandal gap toe |
|
|
Down Syndrome Complications
|
•Intellectual disability: mild to moderate (IQ 35 < 50 < 70)
•Congenital heart disease (50%) • Echocardiogram required @ diagnosis • Most common: endocardial cushion defect (ASD, VSD) •Hearing loss (75%) and otitis media (50-70%) •GI: duodenal atresias (12%), Hirchsprung’s - limited innervation @ distal colon (<1%) •Leukemia (<1%) •Atlantoaxial (neck, cervical spine) instability •Eye diseases: cataracts (15%), refractive errors (50%) •Obstructive sleep apnea (50-75%), •Thyroid disease (15%) |
|
|
causes of down syndrome
|
advanced maternal age
• <30yo: 1 in 1000 • <35yo: 1 in 350 • <40yo: 1 in 85 |
|
|
Prenantal screening for down syndrome
|
•1st trimester: nuchal translucency (ultrasound, checking for space between 2 layers of neck skin), PAPP-A, hCG
•2nd trimester: AFP, Estriol, hCG, Inhibin T21 detection rates: o 1st Trimester = 82-87% o 2nd Trimester = 80% o Combined (integrated) = 95% further testing (to confirm suspected abnormality) o Chorionic villus sampling (CVS): direct placental sample (Miscarriage risk: 1%) o Amniocentesis: miscarriage risk = 1/200 (0.5%) o Non-Invasive Prenatal Testing (NIPT) • Uses cell-free fetal DNA in maternal circulation (present only during specific pregnancy) • Tests for T21, T18, T13, X/Y |
|
|
Non-Invasive Prenatal Testing (NIPT):
|
highly sensitive. Uses cell-free fetal DNA in maternal circulation (present only during specific pregnancy)
• Tests for T21, T18, T13, X/Y; accuracy: • T21: 99.1% • T18: >99.9% • T13: 91. 7% • Fetal gender: 99.4% Can be done as early as the 10th week of gestation. Used as primary screening test only to those at increased risk. If results are abnormal, still recommend confirmatory testing with amniocentesis/CVS |
|
|
Trisomy 18
stats |
oKaryotype: 80% trisomy 18 (nondisjunction event)
oLess severe symptoms → longer survival: • Mosaicism: ~5% • Translocations (partial T18) oFrequency: 1 in 6000-8000 • Second most common autosomal trisomy • Male:Female = 1:4 • Incidence @ 1st trimester: 1 in 400 o High rate of miscarriage (~95%) Live births: • 90% die in first year • 50% die in first 3 months • Average life expectancy: 15 days (pregnancy termination considered more often) |
|
|
trisomy 18 characteristics
|
facial features
• Microcephaly (small head with prominent occiput [posterior head]) • Micrognathia (small chin) • Microphthalmia (small eyes) • Hirsute forehead (excess hair) Body features • Severe growth retardation • Malformed ears • Clenched fingers (overlapping) • Rocker bottom feet • Intellectual disability • Psychomotor delay "Cardinal Features” • Short sternum • Endocrine changes: thyroid, thymus, adrenal • Genitourinary: • Multicystic kidneys • Micropenis, hypoplasia of labia/ovaries |
|
|
Trisomy 13
stats |
o Karyotype: Trisomy 13: 80%
• Most common error: maternal meiosis I • Robertsonian translocation: (13:14): 15% • Mosaicism: 5% oIncidence: 1 in 10,000-20,000 • Male:Female = 1:1 o miscarriage rate: 97.5% Life expectancy: • >90% die within the first year • 80% die in the first month • Average life expectancy: 7 days |
|
|
Trisomy 13
clinical features |
Craniofacial
• Sloping forehead • Deep set eyes • Bulbous nose • Cleft lip and palate -Single central incisor • Malformed ears • Scalp defects (cutis aplasia) •Extremities: polydactyly (abnormal # fingers) •Cardiac: 80% abnormality •Neuro: seizures •Apnea •Holoprosencephaly (66%) • Failed/incomplete separation of forebrain (mostly alobar) Other medical problems: • Feeding difficulties • Seizures (often intractable [difficult to treat]) • Often deaf, blind |
|
|
Sex Chromosome Abnormality Frequency
|
• 45, X: 1 in 2,500-5000
• 47, XXY: 1 in 500 • 47, XXX: 1 in 1000 • 47, XYY 1 in 840 |
|
|
Klinefelter Syndrome
stats |
male with an extra X chromosome
o Karyotype: 47, XXY • Mosaic = 1:5 50% paternal origin, 50% maternal origin Incidence: 1 in 500-1000 Frequent cause of male infertility |
|
|
Klinefelter Syndrome
diagnosis |
• Karyotype/microarray
• Delayed puberty + gynecomastia • During evaluation for infertility |
|
|
Klinefelter Syndrome
clinical features |
•Tall
•Long limbs •↓ facial/body hair (female pattern) •Small genitalia •Gynecomastia (30%): breast development •Decreased testosterone •Replacement starting @ puberty → more masculine physique, improved self esteem •No intellectual disability; however -- • ~10-15% below familial average • Verbal IQ lower than functional IQ •Psychosocial: shy, immature, insecure |
|
|
Klinefelter Syndrome
Treatment |
•Education: expressive language (early intervention up to 3 yrs), individualized academic support
•Puberty: testosterone replacement •Assisted reproductive technology can make child-bearing possible |
|
|
Turner Syndrome
|
females missing X chromosome
o Karyotype: 45, X • 50% due to meiotic non-disjunction (usually, PATERNAL X is lost) • 50% variants: mosaic, deletions/duplications, ring chromosomes Incidence: 1 in 2,000 to 5,000 live female births miscarriage rate (98-99%) ~20% of all 1st trimester miscarriages |
|
|
Turner syndrome clinical features
|
• Prognosis related to amount of X missing
Normal IQ, but typically 10-15 points lower than avg • Also, verbal IQ higher than operational IQ Prenatal: Cervical lymphedema (cystic hygroma), edema Newborn: • Lymphedema of hands/feet • Webbed neck, excess nuchal skin • Shield shaped thorax + increased internipple distance • Congenital heart defects (Bicuspid aortic valve 30%) Childhood: • Short stature • Shield chest + increased internipple distance • Webbed neck • Low hairline Adolescent/adult • Short stature (Avg adult height: 4’11”) • Failure to achieve menarche • Lack of 2o sex characteristics • Infertility (streak gonads) • Cardiac issues: increased risk for hypertension, aortic dilatation and dissection |
|
|
Turner syndrome
Treatment |
•Short stature: growth hormone (as early as 2yo) Average gain: ~2”
•Puberty: estrogen •Echocardiogram (yearly if heart defect, every 3-5 years if structurally normal) •Counsel regarding possible infertility |
|
|
Other common sex chromosome aneuploidies
|
45, X/46, XY mosaic
•Findings similar to Turner syndrome •+ Ambiguous genitalia •Action: remove any testicular tissue •Risk of gonadoblastoma (esp. if in abdomen) 47, XXX (Triple X syndrome) •Only mild → no unusual effects •X inactivation of 2 of X chromosomes 47, XYY •May be taller, but typically no other physical features •Increased risk of learning disabilities |
|
|
Errors in mitosis/meiosis: Non-disjunction
|
• Trisomy: An extra chromosome
• Monosomy: A missing chromosome • Heterodisomy is an error in Meiosis I • Isodisomy is an error in Meiosis II • Mosaicism: postzygotic change where some of the cells are normal and some are abnormal |
|
|
Chromosome translocation
Definition |
• chromosomal abnormality caused by rearrangement of parts between non-homologous chromosomes
• two types: reciprocal (non-Robertsonian) vs Robertsonian • two types: balanced (even exchange of material with no genetic info extra or missing) vs. unbalanced (duplications/deletions occur) |
|
|
Balanced reciprocal translocation
|
• Because there is no gain/loss of genetic information, individuals w/balanced chromosomal translocations are usually phenotypically normal. (occurs in 1/625 individuals)
• Risk to offspring: may lead to malsegregation during meiosis b/c a balanced translocation carrier can produce gametes with unbalanced karyotypes: |
|
|
Unbalanced reciprocal translocation
|
• Unbalanced structural abnormalities usually result in mental retardation & congenital abnormalities in fetus or early pregnancy loss
|
|
|
meiosis of translocation
2 possible pairings |
Adjacent 1 segregation
Adjacent 2 segregation Alternative segregation |
|
|
Adjacent 1 segregation
|
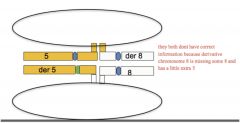
each daughter cell receives one normal and one translocated chromosome
aneuploid gametes (die), each gamete gets one normal and one translocated chromosome (involves del/dup) |
|
|
Adjacent 2 segregation
|
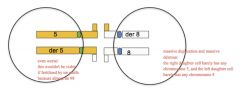
each daughter cell receives one normal and one translocated chromosome (MASSIVE Duplication and deletion)
aneuploid gametes (die), each gamete gets one normal and one translocated chromosome (involves del/dup) |
|
|
Alternative segregation
|
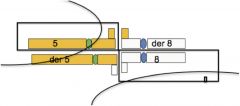
both translocated chromosomes go to one cell and both normal chromosomes go to other cell:
euploid gametes, ½ gametes get both normal chromosomes, ½ get both translocated |
|
|
Three types of chromosomes
|
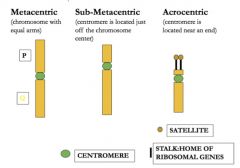
***acrocentric chromosomes: 13, 14, 15, 21, 22***
|
|
|
Robertsonian Translocation
|
when long (q) arms of two acrocentric chromosomes fuse at the centromere, with loss or partial loss of two short (p) arms (p arms carry nonessential genes)
translocation results in a change in chromosome number in normal diploid cells from 46 to 45 •acrocentric chromosomes: 13, 14, 15, 21, 22 •there is no loss of significant genetic information •phenotypically normal but are carriers of Robersonian translocation •most common occurs between 13 and 14 Risk to offspring: homologous acrocentric chromosomes -> disomic gametes (have 2 copies of chromosome) => trisomic conceptions •Translocation Down Syndrome: mother with Robertsonian translocation involving chromosome 21 has 10-15% chance of having baby with translocation Downs |
|
|
Chromosomal deletions
|
syndrome caused by chromosomal deltion spanning several genes that is too small to be detected under microscope using conventional cytogenetic methods - can use FISH or other methods of DNA analysis to identify deletion
caused by errors in crossover during meiosis • chromosomal translocations • crossover within a chromosomal inversion • breaking without rejoining |
|
|
consequences of chromosomal deletions
|
• large deletions usually fatal, small deletions less likely to be fatal
• haploinsufficiency - loss of half of normal activity of a protein causes disease |
|
|
Digeorge (AKA 22q11)
|
syndrome is a deletion syndrome
|
|
|
chromosomal abnormality: duplication
|
also called segmental aneuploidy
causes: • unequal crossing over between two homologous chromosomes or between two chromatids • consequence of crossing over in inversion loop during meiosis |
|
|
Charcot Marie Tooth Disease CMT1A
|
• the most common inherited peripheral neuropathy
• slowly progressive atrophy of distal muscles, progressive weakness • due to DNA duplication in region p11.2-p12 of chromosome 17, - the PMP22 gene that encodes for a myelin protein is duplicated in this region |
|
|
Chromosomal abnormality: Inversion
|
breakage and rejoining of segments within the chromosome
Paracentric inversions and Pericentric inversions |
|
|
Paracentric inversion
|
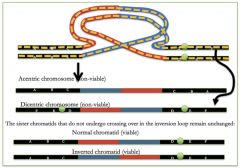
breakage and reunion within the same arm (abnormal gametes that are NOT viable b/c crossing over produces acentric or dicentric chromosome)
|
|
|
Pericentric inversions
|
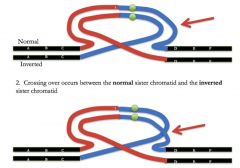
breakage and reunion on either side of the centromere (will produce abnormal offspring - will be alive because there will be one centromere per chromosome, BUT there will be duplications and deletions!)
|
|
|
Chromosomal Abnormalities in Cancer
|
chromosomal rearrangements, deletions, duplications, and inversions have all been implicated in carcinogenesis
|
|
|
Categories of genetic disorders
|
-chromosomal
-microdeletion/duplication -single gene -epigenetic -multifactorial |
|
|
Total Human Genome
|
-3000 megabases in length
-Average gene = 30,000 base pairs -1-2% of the sequence codes for ~20,000 genes -chromosomes 13, 18, and 21 are least dense ->50% of genome = repeat sequences |
|
|
Microarrays
|
•Microarrays are small, solid supports onto which the sequences from thousands of different groups of genes are immobilized, or attached, at fixed spots in a REGULAR pattern.
•The supports: glass microscope slides, silicon chips, or nylon membranes. •The spots: DNA, cDNA, or oligonucleotides. |
|
|
Basics of Microarrays
|
•A microarray has thousands of spots (“array elements”)
•Each spot has millions of copies of a particular strand of DNA representing groups of genes. •In the case of array comparative genomic hybridization, small areas of the chromosome are each assigned a unique spot. |
|
|
Interpreting Spots: Red versus Green
microarrays |
Equal amounts of Patient and Control DNA are
co-hybridized onto a chip and then analyzed! green= deletion red= duplication black= equal ratio do not detect balanced translocations |
|
|
THE RESULTS OF aCGH: FOUR POSSIBILITIES
|
•DIAGNOSTIC (PATHOLOGICAL COPY NUMBER VARIANT)
•UNCERTAIN (? BENIGN COPY NUMBER VARIANT VERSUS SIGNIFICANT) •BENIGN POLYMORPHISMS (COPY NUMBER VARIANT WELL-KNOWN IN THE GENERAL POPULATION) •NORMAL |