Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
248 Cards in this Set
- Front
- Back
|
Bioenergetics
|
Describes the transfer and utilization of energy in a biological system.
|
|
|
change in free energy (∆G)
|
exergonic: energy is released, ∆G is negative, spontaneous
endergonic: energy is added, ∆G is positive, will only proceed if required energy is supplied ∆G0 does not predict the behavior of reactions under physiological conditions because the reactant and product concentrations are rarely close to 1mM in a cell. |
|
|
net ∆G
|
the additive ∆G's for a pathway determine the overall ∆G. if the sum of the ∆G's in a pathway is negative the pathway can proceed even if some of the reactions are endergonic
|
|
|
adenosine triphosphate (ATP)
|
*high-energy phosphate compound*
consists of a molecule of adenosine (adenine +ribose) to which three phosphate groups are attached ∆G0 for hydrolysis of ATP =-7300 cal/mol |
|
|
Most ATP is produces via _________ metabolism in the __________.
|
aerobic, mitochondria
|
|
|
intermediary metabolism
|
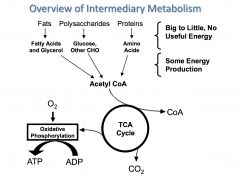
the enzyme-catalyzed process within cells by which energy is extracted from nutrient molecules and that extracted energy is used to construct cellular components
|
|
|
a cell can be described thermodynamically as:
|
an isothermal open system
that perpetuates and replicates itself by means of LINKED organic reactions the most essential type of linkage is between exergonic and endogonic reactions. These do NOT have to be adjacent reactions in common pathways because compounds that contain high-energy bonds (e.g. ATP) are used to transfer energy between non-adjacent reactions |
|
|
the net reaction in aerobic biological systems is the oxidation of substrates. However...
|
few reactions actually involve the direct transfer of electrons from reduced substrates to oxygen. instead, a series of coupled redox reactions occur in the mitochondrial respiratory chain
|
|
|
REDOX reactions
|
Reduced: gains electrons, gains bonds to hydrogen (or loses bonds to oxygen)
Oxidized: loses electrons, gains bonds to oxygen (or loses bonds to hydrogen) LEO the lion says GER |
|
|
Complete oxidation of glucose yields _______
Complete oxidation of palmitic acid yields ________ Why? |
glucose: 15.6 kJ/g (4 Cal/g)
palmitic acid: 38.9 kJ/g (9 Cal/g) b/c carbons in fat are more highly reduced than the carbons in glucose so they contain more potential energy release. (glucose C6H12O6; palmitate C16H32O2) |
|
|
respiratory quotient
|
ratio of CO2 produced to O2 consumed
1.0 for glucose .7 for palmitic acid .82 for albumin |
|
|
Two forms of energy storage
|
chemical and electrical
|
|
|
Chemical energy
|
high-energy phosphate bonds
ATP is the main storage molecule in most cells Others: guanosine triphosphate (GTP) for protein synth uridyl triphosphate (UTP) for nucleotide activation creatine phosphate- energy storage in muscle. synth by creatine phosphokinase (CPK) |
|
|
electrical energy
|
nicotinamide adenine dinucleotide (NAD+/NADH) is the main carrier for electrical energy produced by reduction-oxidation reactions that are part of catabolism
Nicotinamide adenine dinucleotide phosphate (NADP+/ NADPH)- same structure as NAD+/NADH except for addition of a phosphate. a carrier for electrical energy used for anabolic reacts such as fatty acid elongation and nucleotide synth. Flavin adenine dinucleotide (FAD/FADH2)- stores less electrical energy than NADH or NADPH |
|
|
most cells derive energy from the ______. A notable exception being ____ which derive their energy from _____.
|
mitochondria
RBCs glycolysis |
|
|
mitochondrial structure and enzymatic activity
|
OUTER MEMBRANE: some enzymatic activities. relatively permeable.
-----intermembrane space------ INNER MEMBRANE: shuttles for reduced electron carriers, ATP, pyruvate, citrate... transhydrogenase. CATI and CATII. F0F1 ATP synthase. Electron transport and oxidative phosphorylation. 70% protein MATRIX: pyruvate dehydrogenase complex, citric acid cycle, fatty acid oxidation enzymes, urea cycle, genetic component (DNA, transcription, translation) |
|
|
Three general stages of mitochondrial energy production
|
1. Generation of an activated 2 carbon fragment, acetyl-CoA. sources are amino acids, carbohydrates and fatty acids
2. citric acid cycle, which generates reduced electron carriers (NADH, FADH2) 3. Oxidative phosphrylation |
|
|
The respiratory electron transport chain
overview |
Purpose: Conversion of "electrical" energy into "chemical" energy
Location: inner mitochondrial membranes What goes in/ What comes out: electrical energy stored in NADH and FADH2 is converted to chemical energy storage in the from of ATP. Regulation: availability of NADH, FADH2, and ADP; uncoupling proteins |
|
|
the electron transport chain components and steps
|
organized into 5 complexes (I, II, III, IV, and V)
Step1: two electrons passed from NADH to complex I (aka NADH dehydrogenase). for each e-, one hydrogen ion is pumped from matrix to intermembrane space Step 2: the 2 e- are transferred to ubiquinone (aka coenzyme Q) which transfers the 2e- to complex III which transfers them to cytochrome c (2 H+ pumped out) Step 3: cytochrome C brings the 2e- to complex IV. 8 e- interact with O2 and 8H+. 2H2O formed and 4H+ get pumped out of matrix Step 4: H+ gradient used by ATP synthase (aka complex V) to catalyze the synthesis of ATP from ADP+Pi |
|
|
all constituents of the electron transport chain are ____ with the exception of ____
|
proteins
coenzyme Q |
|
|
ATP synthesis is a ______________ mechanism
|
chemiosmotic
|
|
|
1 molecule of NADH through the electron transport chain:
|
3 molecules of ATP
42% efficient: oxidation of NADH ∆G= -220 kJ/mol ATP synthesis ∆G=91.5kJ/mol the other 58% of the energy results in heat generation |
|
|
FADH2 vs. NADH in oxphos
|
FADH2 is less highly reduced than NADH
donates its e- to complex II (aka succinate dehydrogenase). b/c e- from reduced FADH2 enter after complex I, only 2 ATP are produced |
|
|
regulation of the electron transport chain
|
availability of NADH and FADH2
ADP is an allosteric regulator (high ADP increases activity and vice versa) uncoupling proteins UCPs |
|
|
uncoupling proteins (UCPs)
|
create a proton leak allowing protons to flow down their gradient without ATP being produced.
heat is produced UCP1 is known as thermogenin-- brown fat in mammals. humans have other UCPS might be involved in heat production and energy balance and in pathophysiology of obesity and metabolic syndrome. |
|
|
toxins as inhibitors of the respiratory chain
|
aspirin or dinitrophenol short circuit the proton gradient across the inner mitochondrial membrane.
other inhibit components of the chain: rotenone-- complex I cyanide and azide -- complex IV oligomycin -- ATP synthase |
|
|
anaerobic vs aerobic metabolism balance sheet
|

20 fold increase in metabolic efficiency when glucose is metabolized aerobically
|
|
|
why does muscle need an alternative energy storage mechanism?
|
single muscle fiber has up to 15 billion filaments. each filament uses ~2500 ATP/sec when contracting. far in excess of the capacity of a muscle cell to store ATP.
stored instead in another compound: phosphocreatine |
|
|
creatine phosphokinse CPK
|
synthesizes phosphocreatine by catalyzing the reaction of ATP + creatine --> ADP + phosphocreatine
multiple forms of CPK. B (brain type) M (muscle type). dimers BB expressed in all tissues, MM highly expressed in skeletal muscle. MB expressed at relatively higher levels in cardiac muscle (used as a marker of myocardial damage |
|
|
How does phosphocreatine (aka creatine phosphate) work?
|
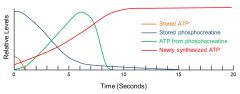
anaerobically donates a phosphate to ADP. levels of phosphocreatine are about 6 times higher than the muscle cell's ATP storage allowing it to be used for ATP source until synthesis catches up
|
|
|
where does the skeletal muscle derive energy from over the duration of activity
|
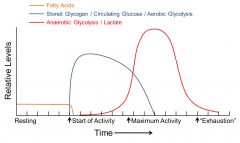
resting muscle: fatty acid metabolism
onset: circulating glucose and glucose derived from stored glycogen (aerobic) maximal activity: oxygen requirements exceed delivery- anaerobic glycolysis predominates. Cori cycle critical for recycling lactate |
|
|
complex III deficiency
|
disruption of the electron transport chain leads to lactic acidosis as a consequence of a shifting to anaerobic metabolism.
|
|
|
mitochondrial genetics
|
some mitochondrial proteins are coded for in mtDNA
mtDNA codes for 13 of the 100 proteins that make the respiratory complexes I,III,IV, and V. maternal inheritance of mitochondria mitochondrial diseases surprisingly common b/c of relatively high rate w/which mutations occur in mtDNA |
|
|
cyanide and carbon monoxide poisoning in the electron transport chain
|
cytochromes of the electron transport chain contain heme as a cofactor. aerobic production of ATP is susceptible to the effects of CN and CO
|
|
|
inherited disorders of the ETC
|
leber's hereditary optic neuropathy- rare form of blindness. mutation is complex I protein
MELAS syndrome- mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. Mutation affects the mitochondrial leucine tRNA in most cases Congenital lactic acidosis- many different mutations, usually affecting complexes III and IV, can present in this manner |
|
|
Glycolysis
overview |
Purpose: glucose metabolism to support energy production. intermediates can be used for other metabolic pathways (lipid- synthesis, amino acid metabolism).
Location: Cytoplasm. all cells What goes in: glucose, or other carbohydrates at intermediate steps What comes out: pyruvate ( for conversion to acetyl CoA if oxygen is available- aerobic metabolism) or lactate (if oxygen is not available -anaerobic metabolism). 2ATP and 2NADH note: only 2ATP if going to lactate because use your 2NADH in the pyruvate-->lactate |
|
|
Glycolysis diagram
|
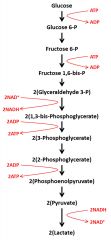
|
|
|
Glycolysis energy investment stage
|
two phosphorylation steps use 1 ATP each
-Glu to G-6-P -Fru-6-P to Fru1,6-bis-P |
|
|
energy generation stage
|
after Fru 1,6-bis P is cleaved to two-carbon fragments. the two carbon molecules are metabolized to 2 pyruvate molecules resulting in the gross total production of 4 ATPs total
|
|
|
Aerobic glycolysis
|
NADH produced from NAD+ at the glyceraldehyde 3-P to 1,3 bis-phosphoglycerate step is used for oxidative phosphorylation. the pyruvate is shuttled into mitochondria for acetyl CoA production and utilization in the citric acid cycle
|
|
|
Anaerobic glycolysis
|
When oxygen availability is low
-pyruvate is converted to lactate. lactate circulates to the liver and is converted back into glucose and recirculates (Cori cycle) -NADH generated by glycolysis is converted back to NAD+ as pyruvate is converted to lactate. this provides energy to the cells producing the lactate |
|
|
four regulatory enzymes of glycolysis
|
Hexokinase
Glucokinase PFK-1 Pyruvate Kinase |
|
|
hexokinase
|
in all tissues
low Km inhibited by Glu-6-P |
|
|
Glucokinase
|
in the liver
High Km upregulated by insulin and downregulated by glucagon |
|
|
phosphofructosekinase -1 (PFK-1)
|
converts fructose 6-P to fructose 1,6-bisP
inhibited by ATP and citrate in the liver- stimulated by insulin and down regulated by glucagon and epinephrine (which are mediated by fructose 2,6-bisP, mediated by PFK-2) |
|
|
PFK1 and PFK2
|
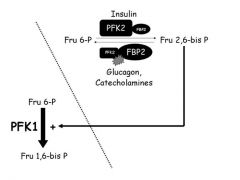
dual function enzymes (two separate catalytic domains on the molecules)
PFK2 (with insulin) is dephosphorylated and catalyzes the reaction of Fru 6-P --> Fru 2,6 bis P *** Fru 2,6 bis P the upregulates PFK1 to catalyze Fru 6-P --> Fru 1,6-bis P *** PFK2 (with glucagon or catecholamines) acts as a phosphatase and catalyzes Fru 2,6 bisP ----> Fru 6-P |
|
|
pyruvate kinase
|
converts phosphoenolpyruvate to pyruvate
activated by insulin and inactivated by glucagon and epinephrine. allosterically inhibited by ATP. |
|
|
is glycolysis an anabolic or a catabolic pathway?
|
in LIVER it behaves as an ANABOLIC pathway. it is stimulated under fed conditions and insulin activates it, so that it can be used for ATP generation
in other tissues, it is used for energy generation at all times, so it behaves as a catabolic pathway |
|
|
regulation by phosphorylation
|
in general insulin leads to dephosphorylation of enzymes and counter regulatory hormones lead to phosphorylation of same enzymes
|
|
|
lactic acidosis
|
accumulation of lactate in the circulation.
occurs when oxygen availability is low. this can occur as a result of decreased perfusion of a tissue (heart attack, hemorrhage), low oxygen tension in the blood (anemia, lung disease), impaired mitochondrial function (aspirin poisoning) |
|
|
during vigorous exercise, lactate can accumulate in muscle
|
lowers the intracellular pH, which may contribute to tonic contractions (muscle cramps)
|
|
|
lactate dehydrogenase (LDH) clinical relevance
|
catalyzes the conversion of pyruvate to lactate
exists in different quaternary structures in different tissues. its levels in blood are used as an indicator of tissue damage. by looking at different isoforms, the affected tissue can be identified |
|
|
Arsenic poisoning
|
lightheadedness progressing to delerium and, ultimately, seizures and death.
arsenic competes with inorganic phosphate as a substrate for glyceraldehyde 3-phosphate dehydrogenase. MOST importantly, it inhibits pyruvate dehydrogenase |
|
|
pyruvate kinase deficiency
|
inheritable hemolytic anemia.
mature erythrocytes lack mitchondria so depend on pyruvate oxidation to maintain cell surface ATP pumps. defective pyruvate kinase causes them to lose their shape and greater tendency toward hemolysis |
|
|
Pyruvate oxidation
|
Purpose: Acetyl CoA formation, catalyzed by pyruvate dehydrogenase (PDH)
Location: Mitochondrial matrix. Pyruvate must be transported to mitochondria for PDH to act on it. What goes in: Pyruvate What comes out: Acetyl CoA (the fuel for the citric acid cycle) Regulation: feedback inhibition on PDH by AcCoA and NADH (products). PDH less active when phosphorylated by PDH kinase (PDK) which occurs under energy sufficiency (ATP). Insulin stimulates dephosphorylation by its phosphatase |
|
|
PDH deficiency
|
inborn metabolic disease: poor growth, muscle weakness, metabolic crises involving hypoglycemia and lactic acidosis.
see also arsenic poisoning for PDH inhibition in brain cells |
|
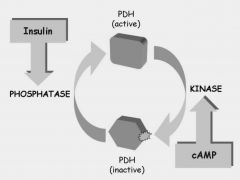
|
PDH regulation
|
|
|
Tricarboxylic acid
|
Purpose: 1. required for the transfer of energy from acetyl CoA to high energy compounds that drive ATP production 2. Intermediate steps of the cycle are tied into other metabolic pathways (gluconeogenesis, urea cycle, heme biosynthesis, amino acid metabolism)
What goes in: Acetyl CoA What comes out: 3 NADH, GTP, FADH2, 2CO2 Regulation: NADH inhibits. ATP inhibits PDH thus limiting substrate |
|
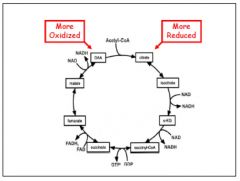
|
TCA cycle graphic
|
|
|
Gluconeogenesis
|
Purpose 1. Provides glucose after 10-18 hours of fasting when liver glycogen is depleted 2. Maintenance of hepatic glucose production for function of organs for which glucose is obligatory (brain and exercising muscle)
Location: liver, mitochondria and cytosol What goes in: Glycerol (from lipolysis), Lactate (from muscle Cori cycle), Glucogenic amino acids (from muscle breakdown) What comes out: glucose Regulation: stimulated by counter-regulatory hormones (glucagon, epinephrine). regulation loci is pyruvate kinase phosphorylation and PFK2/FBP2 |
|
|
steps in gluconeogenesis
|
1. Committed step
conversion of pyruvate to phosphoenolpyruvate (PEP). Pyruvate ---(pyruvate carboxylase)---> Oxaloacetate ---(PEP carboxykinase, PEPCK)---> PEP 2. PEP enters reversed glycolysis until formation of fructose 1,6-bisphosphate. dephosphorylated by Fru1,6bisphosphatase (inhibited by Fru 2,6 bisphosphate) **key regulatory mechanism by insulin and counter reg hormones** 3. hepatic glucose produced by dephosphorylation of Glu-6-P by glucose-6-phosphatase |
|
|
Pyruvate Carboxylase
|
is a mitochondrial biotin containing enzyme that converts pyruvate to oxaloacetate, OAA
OAA is converted to malate to transport out of mitochondria and is then converted back to OAA int he cytosol |
|
|
PEPCK - phosphoenolpyruvate carboxykinase
|
cytosolic enzyme in liver. regulated at the transcriptional level by insulin and catecholamines
|
|
|
major regulatory points for gluconeogenesis
|
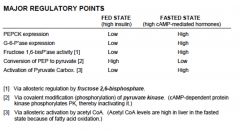
|
|
|
Energy balance for gluconeogenesis
|

2 pyruvate --> 1 Glucose
requires: 4 ATP 2 GTP 2 NADH |
|
|
woman in hospital with pneumonia. unable to eat. you start an IV solution with 5% dextrose
what is the fate of the dextrose? will it make her blood sugar really high? |
1. dextrose --> ATP production in variety of tissues and cell types. macromolecule synthesis
2. normal blood sugar because insulin sensitive tissues will increase uptake and utilization |
|
|
glycogen in the fed state
|
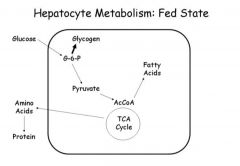
in liver- net glucose uptake. glucose is used for glycogen synthesis and triglyceride synthesis.
in skeletal muscle- insulin promotes glucose uptake and glycogen synthesis. skeletal glycogen is primary means by which blood sugar is kept in the normal range |
|
|
Why Glycogen?
|
High density
Low osmolar load Synthesis and breakdown highly regulated. |
|
|
Fasted state glycogen
|
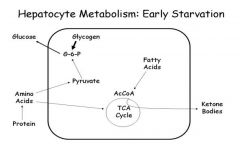
short term maintenance of blood glucose levels
|
|
|
glycogen structure
|
-highly branched
-core primer protein GLYCOGENIN -molecular weight 3,000,000 Da -exists as dense particles -glycogen has a dual purpose: glucose storage depot but also structural scaffold for the enzymes that regulate glycogen metabolism |
|
|
biochemical structure of glycogen
|
-straight chain alpha-1,4 linkage
-branches, alpha-1,6 -1,4 linkages are helical -limit dextrin is when the branch is digested down to the alpha 1,6 branch point |
|
|
glycogen metabolism overview
|
Purpose: acute storage of glucose. release of G6P from glycogen is used locally in most tissues. liver breaks down glycogen for glucose EXPORT. skeletal and cardiac muscle use glycogen as a store for energy for muscle contraction
Location: cytosol in virtually all cells What goes in/ comes out: GLUCOSE Regulation: allosteric reg. by metabolites and calcium. hormonal reg by cAMP-mediated hormones and insulin |
|
|
primary cause of hyperglycemia in adult onset diabetes
|
impaired skeletal muscle glucose disposal as glycogen.
|
|
|
Glycogenolysis
|
shortened by phosphorolysis catalyzed by PHOSPHORYLASE. phosphorylase is specific for alpha 1,4 glycosidic bonds. stops 3 or 4 glucose residues before a branch point.
reaction is isoenergetic K~1.0. reaction is reversible in a test tube phosphorylase is a multisubunit enzyme (heterodimer/heterotetramer). regulated between active and inactive forms in response to hormones. reversible phosphorylation. |
|
|
sequence of reactions required for glycogen degredation
|
1. Phosphorolysis, catalyzed by phosphorylase --> 8 glucose molecules
2. Transferase, catalyzed by debranching enzyme moves 3 glucose molecules from one branch to another 3. Alpha-1,6 glucosidase, also catalyzed by debranching enzyme (bifunctional enzyme) releases one glucose. results in straight chain that can be degraded by phosphorylase |
|
|
glycogenesis
|
initiation: glycogenin functions as an anchor- and it autocatalyzes the formation of initial chain of 6-10 glucose molecules
nucleotide activation: rate limiting enzyme is GLYCOGEN SYNTHASE. substrate is not glucose but phosphorylated glucose (nucleotide-activated glucose) UTP+G1P--->UDP Glu (via UTP-G1P transferase) UDP Glu +Glycogen (n) ---> Glycogen (n+1) (via Glycogen Synthase) |
|
|
Five principles of Regulation of Glycogen Metabolism
|
1. Tissue specificity of regulatory mechanisms
2. Amplification of a hormone signal 3. Organization into a functional unit (the glycogen particle) 4. Multiple control points (reversible phosphorylation) 5. Reciprocal regulation of synthase and phosphorylase |
|
|
Liver versus muscle glycogen metabolism
|

|
|
|
Hormonal regulation of glycogen metabolism
|
synthase and phosphorylase are the focal points of regulation via reversible phosphorylation
-phosphorylase is ACTIVE when PHOSPHORYLATED -synthase is INACTIVE when phosphorylated -both are substrates for phosphorylase kinase which is ACTIVE when PHOSPHORYLATED -All three are substrates for type 1 protein phosphatase (PP1), a major mediator of insulin action -phosphorylase is NOT a substrate for PK-A (cAMP-dependent protein kinase) |
|
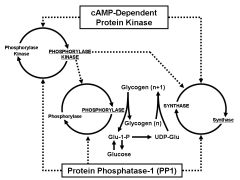
|
Phosphorylase: Glycogen bound, dimer>>tetramer, 1-phosphorylation site, inhibited by glucose in the liver and activated by AMP
Synthase: glycogen-bound, monomer, 9 phosphorylation sites, G6P-activates Phosphorylase kinase: glycogen bound, heterotetramer, multiple phosphorylation sites, Ca++ activates |
|
|
regulation of glycogen synthase
|
9 phosphorylation sites that are substrates for at least 7 kinases
critical sites: 3a-c, 4 are substrates for GSK-3 PK-A 1a, 2a LIVER synthase DOES NOT HAVE cAMP-dependent kinase site |
|
|
Regulation of glycogen synthase by insulin
|
two pathways:
1. Inactivation of GSK-3 (GSK3 normally inhibits synthase) 2. Glycogen bound PP1 (which is a phosphatase for synthase (activates), phosphorylase (deactivates), phosphorylase kinase (deactivates)) |
|
|
co-localization of glycogen
|
glycogen particles are associated with microsomal membranes . They contain the enzymes of glycogen metabolism:
synthase phosphorylase debrancher brancher phosphorylase kinase type-1 phosphatase |
|
|
Flash activation
|
experimental addition of calcium and ATP to intact particles results in activation of phospholyase kinase and phosphorylase. end when ATP is out- indicating functions PP1. analogous to what happens in muscles
|
|
|
regulators of phosphorylase
|
activation:
Glycogen, AMP, ser-14P, inactivate: GLUCOSE inhibition: ATP, G6P, UDP Gluc |
|
|
Type I Glycogen Storage Disease
|
LIVER
Heptomegaly, Growth failure, Hypoglycemia, Lactic acidosis, lipedemia |
|
|
Type II Glycogen Storage Disease
|
All organ w/lysosomes
heart failure in infancy and death before 2 (pompe disease) |
|
|
pentose phosphate pathway
aka hexose monophosphate (HMP) shunt |
Purpose:
-oxidative reactions production of NADPH for fatty acid biosynthesis, steroid biosynthesis, co-factor for antioxidents -non-oxidative reactions production of ribose 5P for nucleotide synthesis. metabolism of diet-derived 5-carbon sugars Location: cytosol of all cells In: G6P Out: NADPH, ribose 5-P Regulation: G6PD (G6P dehydrogenase) is the rate-limiting enzyme. activated by NADP+ and inactivated by NADPH |
|
|
G6PD Deficiency
|
most common deficiency in the world
-hemolytic anemia from exposure to oxidants (antibiotics, antimalarials, antipyretics, fava beans, infection) |
|
|
fructose metabolism
|
phosphorylation by hexokinase
mostly in the liver by fructokinase --> fructose 1 phosphate cleaved by aldolase B to glyceraldehyde. goes through glycolysis from there |
|
|
galactose metabolism
|
phosphorylated in most tissues by galactokinase--> galactose-1-phosphate
then needs nucleotide activation with UTP --> UDP-Gal and is converted to UDP-glucose by epimerase |
|
|
lactose metabolism
|
synth: ß-galactose linked to glucose catalyzed by lactose synthase. in mammary glands
lactase hydrolyzes and produces glucose and galactose. (deficiency in lactase is lactose intolerance) |
|
|
galactosemia
|
deficiency in galactose 1 phosphate uridyl-transferase
newborn with liver disease and bacterial spesis |
|
|
Mucopolysaccharidoses
|
enzyme disorders
recessive inheritance target gene therapy research affects: appearance, physical ability, organ and system functioning and in most cases mental development |
|
|
Glycoproteins
|
proteins with oligosaccharides covalently bound.
|
|
|
term lipid refers to
|
heterogeneous group of organic compounds that are insoluble in water:
-Fatty acids -Triglycerides -Phospholipids -Steroids -Glycolipids |
|
|
summary image of lipid synthesis
|
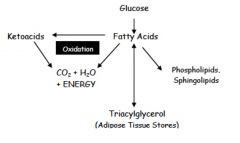
|
|
|
Fatty acid synthesis
|
Purpose: depot for energy storage. precursors for complex lipids
Location: cytoplasm but substrates arise from the cytoplasm. occurs mainly in the liver and lactating mammary glands- adipocytes to a lesser degree IN: Cytosolic acetyl CoA (from mitochondrial citrate) OUT: Fatty acids Regulation: foci is acetyl CoA carboxylase. Fatty acid synthase complex can also be rate limiting |
|
|
Fatty acid oxidation
|
Purpose: provision of energy to most tissues
Location: mitochondria in most tissues. Ketogenesis occurs in liver IN: dietary and stored fatty acids OUT: CO2+water+energy OR ketone bodies exported by the liver regulation: Rate limiting step is transport into mitochondria. indirect regulation of the carnitine shuttle by AcCoA |
|
|
advantages of fatty acids as a long term energy storage
|
-high energy content (9 Cal/g versus 4/cal/g for carbs)
-storage in specialized cells in non-aqueous environment allows for very large stores -depot of substrate for synthesis of structural lipids and signaling compounds |
|
|
structure of fatty acids
|
hydrocarbon chain with a terminal carboxyl group
amphipathic 90% are esterified and associated with lipoprotein particles. unesterified circulate bound to albumin |
|
|
saturation of fatty acids
|
saturated =no double bonds
unsaturated= double bonds (cis is natural formation) trans comes from processing |
|
|
important chain length names of fatty acids
|
palmitic: 16 carbon
linoleic: 18 carbon 2 double bonds arachidonic: 20 carbon 4 double bonds |
|
|
nomenclature notes on fatty acids
|
alpha carbon is the one which the carboxyl group is attached to
beta carbon is the next one omega-carbon is the farthest (terminal methyl carbon). double bonds are counted from the omega |
|
|
Fatty acid synthesis
step 1 |
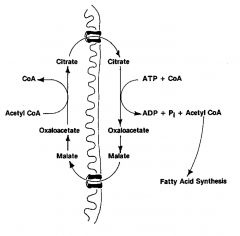
Acetyl group shuttle:
transfer of acetyl CoA from inside the mitochondria to the cytosol. accomplished by the shuttling of citrate b/c AcCoA can't pass through membrane. |
|
|
Malonyl CoA Synthesis from Acetyl CoA in fatty Acid Synthesis
step 2 |
Citrate ------------> AcCoa (via ATP-citrate lyase)
AcCoa ---------> Malonyl CoA (via Acetyl CoA Carboxylase) AcCoa ---------> Malonyl CoA *RLS* regulated by insulin (activation) and counter -regulatory hormones (inactivation) BIOTIN prosthetic group |
|
|
FA Synthesis from malonyl CoA plus Acetyl CoA
Step 3 |
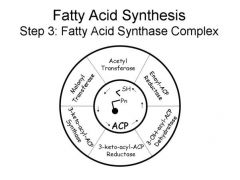
catalyzed by fatty acid complex
reduction reaction with NADPH combines AcCoA (2carbon) and malonyl CoA (4 carbon) = 6 carbon fatty acid |
|
|
cytosolic
consists of 7 enzymes central to its functioning is ACYL CARRIER PROTEIN ACP uses pantothenic acid (vitamin B5) for binding of malonyl CoA |
|
|
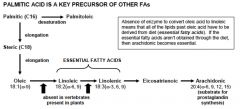
Palmitic acid is a Key precursor of other fatty acids
which acids are essential fatty acids? |
|
|
|
Elongation of Fatty Acids Beyond C16
|
Occurs in the endoplasmic reticulum and in mitochondria
production of very-long-chain fatty acids (up to 24 carbons) occurs in brain and some other select tissues like the adrenal gland |
|
|
Desaturation of fatty acids
|
occurs in the endoplasmic reticulum
catalyzed by mixed-function oxidases fun fact: linoleic and linolenic acids are essential because body lacks oxidases than can introduce double bonds past carbon 9 |
|
|
Triacylglycerol synthesis
|
occurs in liver and adipose tissue
adipose- used from energy storage liver- fatty acids are exported as VLDL for use by other tissues. |
|
|
Fatty acid oxidation
step 1 |
HORMONE-SENSITIVE LIPASE
-hydrolyzed triglycerides, releases free fatty acids from adipocytes -triglycerides are converted to monoglycerides then to FFA plus glycerol -glycerol metabolism accounts for 5% of energy from triglycerides cannot be metabolized by adipocytes. exported for glycolytic/neogenic path **hormone sensitive lipase is activated by cAMP-dependent kinase. inactivated by insulin** |
|
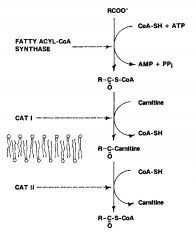
Fatty Acid oxidation: mitochondiral transport
step 2 **CPT aka CAT |
oxidized in the mitochondria outer and inner membrane
chain lengths over 12 carbons require a system to transport them 1. acyl-CoA synthesis 2. CPT I converts fatty acyl S CoA + carnitine to fatty acyl-carnitine + CoA-SH 3. CPT II converts fatty acyl-carnitine + CoA-SH to fatty acyl-S-CoA +Carnitine |
|
|
carnitine
|
since the carnitine transport system is the main point for the hormonal regulation of fatty acid oxidation- only oxidation of long chain fats is subject to regulation
can be obtained from the diet or synthesized in liver from lysine and methionine. mostly stored in SKELETAL muscle |
|
|
carnitine deficiency
|
primary - can't synth or defect in reabsorption
secondary - disordered oxidation in wasting CPT1-deficiency affects liver and causes hypoglycemia CPT-II deficiency most often effects cardiac muscle and skeletal muscle and causes cardiomyopathy or muscle weakness |
|
|
ß-oxidation: Dehydrogenase Step
Fatty acid oxidation step 3 |
Acyl-CoA dehydrogenases catalyze
fatty acyl-S-CoA +ETF_FAD --> enoyl-S-CoA +EFT-FADH2 enzyme is itself reduced. NET SYNTH OF 2 ATP |
|
|
the rest of ß-oxidation
Fatty acid oxidation |
enzymes:
Enoyl-CoA hydratase 2-hydroxyacyl-CoA dehydrogenase acetyl-CoA acetyltransferase results in removal of 2 carbons and the production of acetyl CoA. |
|
|
Energy Balance of palmitate oxidation
|
2 ATP per FADH2+3ATP ner NADH =35 ATP
8 acetyl-COA yield 96 ATP (citric acid cycle plus oxphos) total: 131 ATP for mol palmitoyl-CoA |
|
|
Ketone body formation
|
in the liver produced rather producing ATP from fat metabolism
2 acetyl-S-CoA + H2O --> acetoacetate acetoacetate is further converted to ßOH butrate and to trace amounts of acetone. |
|
|
control of ketogenesis
|
overflow pathway
in fasted state: net flux of oxaloacetate up the gluconeogenesis path. decrease in oxaloacetate results in decreases acetyl CoA utilization for citrate formation. So, acetyl CoA is converted to acetoacetate. HEPATIC KETOGENESIS IS FAVORED WHEN CARBOHYDRATE AVAILABILITY IS LOW. RTL is HMG-CoA synthase that forms acetoacetate. HMG-CoA synthase highest levels in liver. thus major ketogenic organ. Continuously as fuel source by myocardium. in fast brain and muscle use ketone body metabolism. |
|
|
ketone metabolism
|
in the peripheral tissues, ß-OH butyrate is converted to acetoacetate, yielding NADH. acetoacetate then enters TCA cycle
|
|
|
lipid metabolism in diabetes:
|
in the periphery, the lack of insulin is a potent stimulus for lypolysis
triglycerides released and transported to the liver where ketogenesis is fully activated. DIABETIC KETOACIDOSIS!! |
|
|
alpha-oxidation
|
Purpose: metabolism of FA's that cannot under go direct ß-oxidation
Location: peroxisomes In: FA Out: alpha-OH FAs Regulation: substrate driven Refsum's disease (alpha-hydroxylase deficiency): accumulation of phytanic acid derived from chlorophyll. Rx diet |
|
|
omega-oxidation
|
purpose: alternative metabolism of FAs
location: extra-mitochondrial (microsomal) In: FA Out: Dicarboxylic FAs Regulation: substrate-driven clinical note: upregulated if ß-oxidation is defective |
|
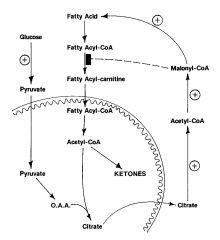
regulation of fatty acid metabolism
|
Primary site of oxidation reg: CPT I (omm)
acetyl CoA carboxylase is the PRIMARY regulatory point acetyl CoA carboxylase is activate via insulin dephosphorylation -promotes fatty acid synth and inhibits oxidation at the mitochindrial transport step -promotes fatty acid synth by increasing the content of ATP citrate lyase and fatty acid synthase. increased production of oxaloacetate from pyruvate provides for fatty acid synthesis and inhibits ketogenesis |
|
|
Medium chain acyl CoA dehydrogenase deficiency MCAD
|
most common fatty acid oxidation disorder
cause of SIDS med chains most abundant in milk - common in infancy. Rx: avoid fats, admin carbs via IV during periods of stress, admin carnitine |
|
|
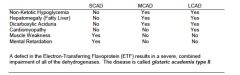
disorders of fatty acid oxidation chart
chain length disorders |
|
|
|
glutaric academia type II
|
electron transferring flavoprotein defect causes combined sever impairment of all the dehydrogenases
|
|
|
cholesterol structure
|
Sources: diet (animal products) and endogenous synthesis (de novo)
Functions: cell membranes, steroid hormones, bile acids Structure: steroid nucleus, hydrocarbon tail, fatty acid tail Lipoproteins transport cholesterol esters (CE) and Triglycerides (TG); CE is the storage form of cholesterol |
|
|
ACAT
|
Enzyme
Acyl CoA Cholesterol Acyltransferase transfers long chain FFA to C3 for cholesterol ester storage in cells |
|
|
Synthesis of cholesterol
|
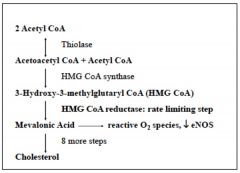
in all tissues
basically putting together a series of acetate molecules. 2 acetyl-COA + acetyl CoA--> HMG CoA --> mevalonic acid by HMG CoA reductase RLS >>>>8steps>>>CHOLESTEROL |
|
|
Regulation of cholesterol synthesis
|
1. HMG CoA Reductase is the major site of regulation
BUT synthesis is also regulated by LIVER enzymes: ex: cytosolic enzyme makes mevalonic acid (req. NADH) 2. Gene expression is controlled by insulin and sterol regulatory element-binding protein (SREBP) 3. low cholesterol levels increase high cholesterol levels inhibit SREBP expression. Increased cholesterol also decreases the protein's stability and phosphorylation of enzyme decreases its activity 4. statin drugs are competitive inhibitors of HMG CoA Reductase |
|
|
Metabolism of cholesterol
|
-50% of free cholesterol in GI tract is reabsorbed and sent back to liver
-cholesterol is converted to bile acid (cholic acid) in liver by 7alpha-hydroxylase. conjugated to form bile salt. secreted in bile -97% of bile acids are reabsorbed. returned through enterohepatic ciculations |
|
|
lipoprotein overview
|
macromolecules made of inner lipid core and outer shell of protein, phospholipid and free cholesterol
Function: carry hydrophobic triglycerides and choesterol esters, fat soluble vitamins, and drugs in plasma to tissues vary in size and density due to compositions |
|
|
apolipoproteins
|
confer structure, receptor binding, and enzyme activation
|
|
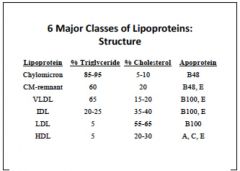
|
CM: largest, least dense, ApoB48, metabolized into chylomicron remnants (chylomicrons are parts of CM's that have lost bulk of TGs and gained protein ApoE)
VLDL: half TG, 20%chol, ApoB100, ApoE, constructed in the liver IDL: metabolic product of VLDL. less TG but more cho, ApoB100, ApoE LDL: metabolic product of IDL, almost completely chol, ApoB100 HDL: almost no TG, but has series of specialized proteins that get swapped around w/other lipoproteins. |
|
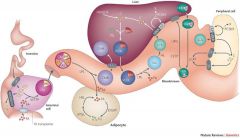
|
3 pathways all together
|
|
|
microsomal triglyceride transport protein
|
packages FA, TGs and ApoB48 into chylomicrons.
|
|
|
Exogenous pathway of lipoprotein metabolism
|
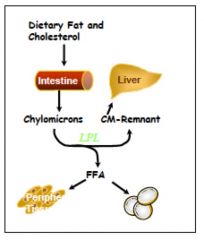
chylomicrons are produced in the small intestine from micelles
secreted into mesenteric lymph and travel through the thoracic duct to the blood. they acquire Apo E and ApoCII from HDL they are metabolized by lipoprotein Lipase (LPL) on capillary cells. ApoCII is the cofactor required for this. ApoE mediates uptake by the liver: autosomal recessive mutations would result in disease states where TG levels are in thousands b/c CM remnants and VLDL cannot be hydrolyzed |
|
|
Endogenous pathway
VLDL --> IDL --> LDL |
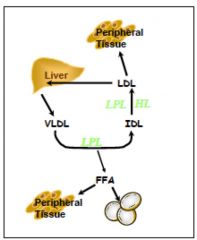
VLDL synthesized in the liver from TG (regulated by free fatty acid influx, alcohol, drugs)
It acquires ApoCII for LPL and APoE for remnant uptake from HDL. modified by LPL in capillaries , removing TG for tissues, becomes IDL IDL is further hydrolyzed by LPL and hepatic lipase (HL). exchanges Triglycerides for cholesterol ester with HDL via Cholesterol Ester Transfer Protein (CETP), becoming LDL |
|
|
Lipoprotein lipase
|
LPL is an extracellular enzyme that is anchored by heparin sulfate to capillary walls esp. adipose, cardiac and skeletal muscle.
activated by ApoCII on chylomicrons, VLDL, IDL to hydrolyze triglycerides into free fatty acids and glycerol. 2 isomers: -adipose enzyme has large Km -cardiac enzym has small Km synthesized in adipose and muscle and transferred to capillary wall. regulated by insulin. activated by fibrate drugs |
|
|
autosomal recessive LPL deficiency
|
rare
severely elevated TG after birth treat by restricting fat in diet |
|
|
ApoCII deficiency
|
autosomal recessive
not as severe as LPL deficiency b/c LPL has some activity w/o ApoCII |
|
|
Cholesterol Delivery (LDL)
|
LDL derived from VLDL after lipase mediated hydrolysis
delivers cholesterol to tissues by ApoB100 binding to LDL receptor-->endocytosis Modified LDL (e.g. oxidized) can also be taken up by macrophages and become FOAM CELLS in endothelial plaques hepatic lipase (HL) also acts on LDL to form small, dense LDL |
|
|
Lipoprotein (a):
|
small and large LDL become small and large Lp(a) formed from linkage of Apo(a) to Apo B-100 via disulfide bond. promotes thrombosis
Apo (a) varies in length determined by genes. variability has an effect of Lp(a) density Lp(a) has been shown to correlate with CVD |
|
|
LDL-receptor
|
LDLR is a glycoprotein that binds ApoB100 and ApoE (NOT ApoB48)
causes endocytosis of LDL particles gene expression controlled by SREBP |
|
|
LDL receptor related protein
|
LRP is a larg complex of 4 LDLR, which binds ApoE-enriched particles (CM and VLDL remnants) but NOT LDL
|
|
|
LDLR mutations
|
familial hypercholesterolemia
many different. can be AD, AR, or compound HZ LDL is really high because receptor mutations prevent LDL from being taken up by cells |
|
|
HDL metabolism
|
particles start as pre ßHDL or nascent HDL made in liver and intestines.
nHDL has many phospholipids and ApoA1 takes up free cholesterol from macrophages and tissues. eventually becomes HDL2. note: along the way it converts free cholesterol to CE using LCAT (enzyme) |
|
|
HDL mature
|
transfers cholesterol to LDL
delivers cholesterol to tissues using SRB1 receptor can be hydrolyzed in the tissues using hepatic lipase to smaller denser HDL that don't deliver cholesterol and get excreted by the kidney |
|
|
reverse cholesterol transport
|
nHDL picks up free cholesterol using transporter ABC1
converts free cholesterol into cholesterol ester via LCAT resulting in mature HDL HDL can now deliver cholesterol to tissues so you can make steroids, to the kidneys to be excreted, or return it to the liver by binding to SRB1 HDL can also swap cholesterol with VLDL particles to make LDL |
|
|
interpreting fasting lipid profiles
|
1. obtain fasting sample to eliminate CMs
2. Total cholesterol and triglycerides are measured directly (TC largely indicates LDL, TG indicate CM and VLDL) 3. HDL is measured directly after precipitation of Apo B100-containing lipoproteins (VLDL< IDL< LDL) 4. LDL in a standard lipid profile is an estimation |
|
|
Estimated LDL
|
Estimated LDL = TC - HDL - (TG ÷5)
TG÷5 approximate VLDL cholesterol ***when TG< 400*** CANNOT USE IF TG≥400 |
|
|
Dyslipidemia of diet and obesity
|
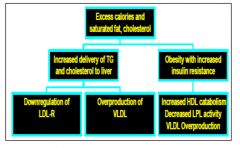
|
|
|
dietary treatment of lipid disorders
|
reduce saturated fat, cholesterol <200 mg/d (decreases TC 10-20%
Fiber, plant sterols inhibit cholesterol absorption Substitution with specific fat sources Control or prevent obesity by caloric restriction and exercise |
|
|
Medical therapy of lipid disorders
|
-Bile acid sequestrants decrease reabsorption
-Cholesterol absorption inhibitors have intracellular action -HMG CoA reductase inhibitors- STATINS upregulate LDL receptors (20-50% LDL decrease) -niacin inhibits VLDL secretion reduces TG via LPL (dec TG, LDL, inc HDL |
|
|
visual for treatments for dylipidemia
|
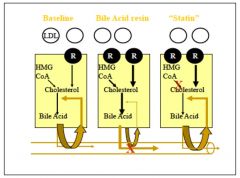
|
|
|
Porphyrin
|
Contain four pyrrole rings connected together to form macrocyclic compound that readily binds iron at its center.
Oxygen carrying Heme is Ferrous Fe2+ 2 ALA makes 1 porphyrin, each containing 2 glycine |
|
|
Heme (Fe2+ Protoporphyrin IX)
|
Most abundant tetrapyrrole in vertebrates
Fe conjugated in center of tetrapyrrole rings binds O2, CO2, CO. CO and CN preferentially bind heme containing cytochromes including ETC cytochrome b-c1 and cytochrome oxidase as well as hemoglobin and myoglobin Heme is responsible for oxygenation of tissues Thalassemia: defective heme due to imbalanced alpha and beta globulin subunits → decreased erythropoiesis and severe anemias |
|
|
Uses of Heme
|
Found in hemoglobin and myoglobin
Cytochromes (electron transport chain ETC, cytochrome P-450) Cytochrome P450 enzymes are present in most tissues of the body, and play important roles in hormone synthesis and breakdown; also function to metabolize potentially toxic compounds, including drugs and metabolic breakdown products such as bilirubin, principally in the liver Enzymes (such as catalase) |
|
|
Discovery of the Biosynthetic Origin of Heme Major Findings
|
All 4 Heme Nitrogen atoms in 4 pyrrole are derived from glycine
Most heme in the blood cells have a half-life of ~ 120 days There is also a small pool of more slowly turned over heme in the body |
|
|
Heme Synthesis-Formation of ALA
STEP 1 **starts in the mitochondria and goes to ALA to leave** |
Glycine and Succinyl CoA provide all of the building blocks of heme synthesis
go to ALA by enzyme ALA synthase RLS |
|
|
Heme is produced in:
|
Liver:
Variable rates for use in cytochrome P450 enzymes Rate is controlled by end-product inhibition of first and rate-limiting step: action of ALA synthase Bone Marrow RBC-producing cells make heme at constant rates matched to globin synthesis Rate controlled by erythropoietin (EPO) and availability of iron |
|
|
ALA synthase is the rate limiting step of heme synthesis.
|
Short Half-life. Most rate-limiting steps are furthest upstream in metabolic pathway as to minimize unnecessary loss of time and resources.
Glucose inhibits as well. Common treatment is to give candy bar for patients with ALA accumulation Stimulated by hypoxia and phenolbarbitol. Drugs that are metabolized by the P450 system consume heme molecules and hemin decreases, which upregulates heme synthesis (needed for P450 system inhibited by hemin (formed when Fe in excess heme is oxidized) -Drugs that are metabolized by the P450 system consume heme molecules and hemin decreases, which upregulates heme synthesis (needed for P450 system |
|
|
Heme Synthesis - Porphobilinogen
STEP 2 |
ALA-->prophobilinogen via ALAdehydrogenase
(cytosol) |
|
|
Porphobilinogen →urophorphyrinogen
STEP 3 (several steps in the cytosol of heme synthesis ) |
First pyrrole is porphobilinogen (porphyrin ring containing 1 nitrogen 2 glycine)
First linear tetrapyrrole is hydroxymethylbilane (4 porphobilinogen, 4 N, 8 Gly) First cyclic tetrapyrrole (porphyrin) is uroporphyrinogen (Photosensitive = can interact with UV Light) **this is followed by more steps in mitochondria that lead to protoporphyrin IX** |
|
|
Heme Synthesis - Incorporation of Fe 2+
STEP 4 |
Fe 2+ is spontaneously introduced into protoporphyrin IX
Rate enhanced by mitochondrial enzyme ferrochelatase |
|
|
lead inhibition in heme synth
|
Ferrochelatase is inhibited by lead → accumulate protoporphyrin IX
ALA dehydratase is inhibited by lead → accumulate ALA |
|
|
Iron Metabolism is closely coordinated with Heme Metabolism
|
Heme is major repository for iron in the body
Excess iron is toxic due to free radical generation Serum iron is complexed to transferrin Cellular iron is complexed to ferritin Excess porphyrins accumulate → porphyrias Dietary Free Iron is poorly absorbed, while heme is absorbed by carnivores. |
|
|
Heme is used in Hemoglobin Synthesis
|
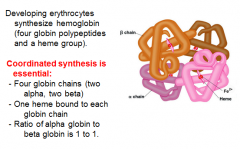
|
|
|
Regulation of Hemoglobin Synthesis
|
Excess globin turns off protein (globin chain) synthesis
Excess heme (oxidized to hemin) turns off ALA synthase In the absence of heme, globin synthesis is halted, b/c heme controlled inhibitor is active and phosphorylated eIF-2 is inactivated. Liver’s cytochrome P450 constantly consumes heme to detoxify harmful substances → upregulation of ALA synthase. De-repress ALA synthase means activate, because its repressor, excess heme, is consumed by P450 for drug detox. |
|
|
HCI (heme controlled-inhibitor) protein regulates hemoglobin synthesis in the presence of heme.
|
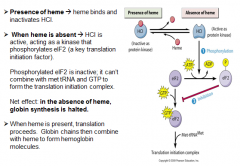
|
|
|
Porphyrias
|
Rare defects in heme synthesis
Autosomal Dominant (except congenital erythropoietic porphyria) inheritance Many heterogeneous mutations in enzymes throughout heme synthetic pathway All porphyrias exhibit decreased heme synthesis → accumulate upstream intermediates prior to block |
|
|
Acute Intermittent Porphyria
|
Acute intermittent episodes of accumulation of porphyrin ring due to inability of 4 porphobilinogen to condense into hydroxymethylbilane.
ALA synthase triggered during low glucose and hypoxic conditions. dark purple urine no photsensitivity ALA still active |
|
|
Porphyria Cutanea Tarda
|
Chronic disease of liver and bone marrow
Uroporphyrinogen III accumulates in the urine Photosensitive Cutaneous symptoms influenced by iron overload, sunlight, Hep B, C, or HIV Dark red urine MOST common porphyria blistery hands |
|
|
Erythropoietic Protoporphyria
|
- ferrochelatase deficiency
-photosensitive -Distinct from congenital erythropoietic porphyria -block in conversion of protoporphyrin IX to heme |
|
|
Porphyria Classification
|
Erythropoietic (cutaneous symptoms, appear in early childhood; 10% complicated by progressive cirrhosis and liver failure)
Acute Hepatic Porphyrias (example: acute intermittent porphyria) Characterized by acute attacks of GI, neurologic/psychiatric, and cardiovascular symptoms .Accumulation of PBG & ALA in urine Attacks triggered by drugs, alcohol, fasting (low blood glucose) Enzyme block decreases production of heme (hemin) – de-repression of ALA synthase. Chronic Hepatic Porphyrias (example: porphyria cutanea tarda) Porphyrin accumulation disorders resulting in cutaneous symptoms associated with liver disease |
|
|
Degradation of Heme
|
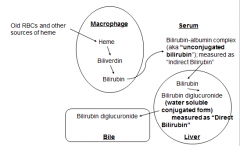
Normal RBC Life Span = 120 days
RBCs are degraded in spleen and liver 85% of heme being degraded comes from mature RBCs, 15% from immature RBCs and extraerythroid hormone Heme is degraded in several steps and excreted in the urine and feces |
|
|
Degradation of Heme from Bile to Excretion
|
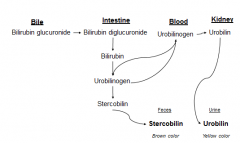
Biliverdin, Bilirubin: Changing colors of a bruise reflect changing patterns of heme degradation intermediates
Urobilinogen (soluble form of Bilirubin) is colorless Stercobilin turns feces brown Urobilin turns urine yellow |
|
|
Solubility in heme degradation and tranport mechanisms
|
Bilirubin is only slightly soluble in plasma
Following heme degradation in spleen, it is conjugated to albumin to deliver it to the liver Hepatocytes glucoronidate Bilirubin → secrete into bile Some Bilirubin → Urobilinogen in intestine |
|
|
Jaundice classifications
|
Hemolytic Jaundice
Hepatocellular Jaundice Obstructive Jaundice Neonatal Jaundice |
|
|
Hemolytic Jaundice
|
Normal excretion capacity of bilirubin is 3g/d. Normal production is 3 mg/day
Excess capacity allows system to handle increased excretion When there is massive lysis of RBC → overwhelm excretion capacity → bilirubin accumulates in plasma. *Sickle cell anemia *Pyruvate kinase deficiency *G6P Dehydrogenase deficiency |
|
|
Hepatocellular Jaundice
|
Damaged hepatocyte decreases liver conjugation of bilirubin
Conjugated bilirubin is inefficiently secreted to bile and diffuses into blood Enterohepatic circulation of intestinal urobilinogen is decreased → more in blood Dark urine, pale stools (remember urobilinogen is colorless) High AST and high ALT Indicates Liver Damage Other symptoms include anorexia and nausea |
|
|
Obstructive Jaundice
|
Obstruction of bile duct
Reduced bilirubin secretion into intestine as bile → regurgitation or backflow of bilirubin into blood Possible causes: live tumor, pancreas head tumor, bile stone Associated symptoms: abdominal pain, nausea, pale, clay colored stools, urine darkens upon standing |
|
|
Neonatal Jaundice
|
At birth, low activity of hepatic bilirubin glucouronyltransferase → accumulate unconjugated bilirubin. Most susceptible are premature new-born babies.
Plasma unconjugated bilirubin overwhelms albumin’s capacity to bind → cross BBB and cause toxic encephalopathy Treated w/ blue fluorescent light to convert unconjugated bilirubin to more polar and water-soluble isomers. |
|
|
Nucleotides
|
building blocks of DNA/RNA
Also components of 2nd messenger systems (cAMP), energy storage (ATP, GTP), and enzymatic cofactors (NAD) Nucleoside: purine/pyrimidine attached to 1’C Purines: A & G; 2 rings Pyrimidines: C, T (DNA), and U (RNA); 1 ring Nucleotide: 1+ phosphates attached to 5’C |
|
|
Nucleotide Synthesis (using PRPP)
|
PRPP (5-phosphoribosyl-1-pyrophosphate) = common precursor pentose sugar + phosphate to which purines/pyrimidines are attached
PRPP synthetase: activated by Pi + ATP, inhibited by purines (End-product inhibition) De novo synthesis: Purines: Purine rings assembled sequentially onto PRPP → purines Pyrimidines: Pyrimidine ring synthesized independently then attached to PRPP when complete |
|
|
Salvage pathways (using existing bases without requiring de novo base synth)
|
Purines: phosphoribosyltransferases attach bases to PRPP
Pyrimidines: bases rarely salvaged |
|
|
Deoxyribonucleotide Synthesis
|
Ribose = substrate for synthesis reactions
When deoxyribonucleotides are needed for DNA synthesis (S-phase in cell cycle), ribonucleotide reductase converts necessary diphosphate ribonucleotides (NDP) by donating own hydrogen atoms => deoxyribonucleoside diphosphates (dNDP) |
|
|
Purine De Novo Synthesis
|
Adenine (a) & Guanine (g))
SUMMARY: Purines assembled directly onto PRPP (multistep synthetic pathway) Individual atoms of purine ring donated by several different compounds Reaction series generate inosine monophosphate (IMP, with hypoxanthine base) Common precursor of both GMP and AMP |
|
|
Purine Metabolic and Salvage Pathway R
|
Recycling pathway for purines obtained from normal turnover or diet
Adenine phosphoribosyltransferase (APRT) converts adenine → AMP Hypoxanthine-guanine phosphoribosyltransferase (HGPRT) converts hypoxanthine → IMP, guanine → GMP |
|
|
Purine Salvage Defects
|
Lesh-Nyhan Syndrome
Caused by defects in HGPRT (nearly complete deficiency, X-linked recessive) Combo → ↑ purine turnover → hyperuricemia Loss of negative feedback → ↑ synthesis UA buildup → ones (kidneys), gouty arthritis (joints), soft tissue deposition, motor dysfunction, cognitive deficits, behavioral disturbances(ex: self-mutilation via lip-biting) |
|
|
Purine Degradation
|
Dietary purines degraded in intestine and don’t contribute much to nucleic acid synthesis
Adenosine deaminase (ADA) converts adenosine → inosine ADA deficiency → accumulation of adenosine other purine degradation disorders: Gout, SCID |
|
|
SCID (severe combined immunodeficiency disease)
|
Lymphocytes have highest ADA activity so most affected by block in DNA synthesis
Manifests with ↓ T and B cells (& NK cells) Death by age 2 without treatment |
|
|
gout
|
Hyperuricemia due to overproduction/underexcretion (more common) of UA
Primary (overproduction) Idiopathic: genetically increased Vmax for PRPP production Lesch-Nyhan: ↑ flux through Purine synthesis pathway Secondary (underexcretion) Idiopathic, due to kidney dysfunction, drugs, lead exposure Symptoms: Deposition of Na+ crystals → joints/soft tissues → Acute (soon chronic) arthritis (classic location: MTP of big toe) → nodular trophi of chronic tophaceous gout in soft tissues Aspirated synovial fluid from joint shows crystals Inflammatory reaction to UA UA kidney stones Rx: Anti-inflammatory drugs: Colchicine, ASA-Don’t affect UA levels, only inflammatory response Increase UA excretion-Probenecid, sulfinpyrazone Decrease UA synthesis -Allopurinol: blocks Xanthine oxidase therefore UA synthesis |
|
|
Pyrimidine Synthesis: Overview
|
Pyrimidine ring synthesized then attached to PRPP (vs. purine ring’s direct assembly)
Formation of carbamoyl phosphate by carbamoyl phosphate synthetase (CPS) II = rate-limiting/regulated step Inhibited by UTP (end product), activated by PRPP/ATP Synthesis requires glutamine, aspartic acid, and PRPP (just like Purine) Orotate = final ring that’s attached to PRPP |
|
|
Carbamoyl Phosphate Synthetases
|

|
|
|
Pyrimidine Synthesis: Cytidine
|
UMP (via pyrimidine synthetic pathway) is phosphorylated → UDP + UTP
Glutamine donates amino group → UTP → CTP |
|
|
Pyrimidine Synthesis: Thymine
|
Thymidylate synthase converts dUMP → dTMP, using N5,N10-methylene-tetrahydrofolic acid as carbon/hydrogen donor
THF (folate) is therefore essential for dTMP synthesis/DNA synthesis |
|
|
Folate Physiology
|
Dietary folate → dihydrofolate (DHF) → tetrahydrofolate (THF, via dihydrofolate reductase or DHFR)
|
|
|
Folate and cancer
|
Methotrexate: DHFR inhibitor used against cancer
→ THF deficiency from DHF ↓ dTMP → purine/DNA synthesis inhibition Also ↓ cancer cell growth rate 5-fluorouracil: anticancer agent/thymidine analog that permanently binds/inhibits thymidylate synthase (blocks dUMP → dTMP) Sulfonamides: competitively inhibit bacterial DHPS req’d for THF synthesis Competes with precursor PABA (para amino benzoic acid) Used in combination with Trimethoprim: inhibits bacterial DHFR |
|
|
folate deficiency
|
Neural tube defects
THF critical in the first few weeks of fetal life Low levels → spina bifida (congenital defect of vertebral column due to defective neural tube closure during embryologic development) and anencephaly (neural tube defect → absence of major portion of brain) Megaloblastic anemia Low THF halts DNA synthesis in bone marrow. B12 required for adequate folate regeneration B12 deficiency → anemia + neurologic findings (which don’t respond to folate supplements) |
|
|
Pyrimidine Salvage and Degradation
|
Rarely recycled from diet or nucleic acid breakdown (easily made in body)
Ring is degraded → highly soluble products However, pyrimidine nucleotides/nucleosides can be salvaged/reused |
|
|
amino acid metabolism overview
|
Excess AA are deaminated and their carbon skeletons are used for energy by entering gluconeogenesis or ketogenesis.
Toxic ammonia groups converted to urea in liver and excreted. |
|
|
The body’s AA “pool”:
|
The pool is supplied by: what you bring in and what you turnover
Absorption from diet (~100g/day) Normal protein degradation (~400g/day) -- recycle AAs Synthesis of nonessential AA from precursors (varies) The pool is used for: Protein synthesis (~400g/day) Synthesis of other products, e.g. epinephrine (~30g/day) Metabolism for energy production (v |
|
|
DIETARY AMINO ACIDS: DIGESTION AND ABSORPTION
|
Dietary protein digestion begins in the stomach.
Pepsinogen converted to Pepsin, an enzyme that begins process of cleaving polypeptides into shorter chains. Digestion continues in the duodenum. CCK:released in response to fats/proteins; Secretin released in response to stomach acid. Both signal the release of pancreatic enzymes. |
|
|
Zymogens:
|
inactive precursor enzymes secreted by the pancreas (into the Duodenum). Examples (zymogen → active enzyme):
Trypsinogen → Trypsin Chymotrypsinogen → Chymotrypsin Proelastase → Elastase Procarboxypeptidase A+B → Carboxypeptidase A+B |
|
|
Enteropeptidase
|
is secreted into the Duodenum in response to food
Activates Trypsinogen → Trypsin Trypsin (very effective protease) then activates all pancreatic zymogens, including more Trypsinogen (autocatalytic). Activated enzymes cleave large polypeptides into shorter oligopeptides and single AAs |
|
|
Aminopeptidase
|
(on intestinal wall) finishes digestion of remaining oligopeptides into AAs, di-, and tri-, peptides
|
|
|
At least 7 different systems exist to transport AA across intestine wall.
|
The same transporters resorb AA in the proximal convoluted tube (PCT) of kidney to keep AA from being excreted in urine.
|
|
|
branched chain AA (BCAA) are
|
are not metabolized by liver; released directly to systemic circulation (usually metabolized in muscle).
|
|
|
absorption disorders
|
cystinuria
Defect in transport of dibasic AA (COAL: Cystine, Ornithine, Arginine, Lysine) Excess cystine in urine leads to kidney stones One of the most common inherited disorders hartnup's disease Defect in uptake of tryptophan and other neutral AA Resembles a dietary deficiency of Niacin, called Pellagra (Tryptophan can be converted into Niacin) Results in 3 “D”s: Dermatitis, Diarrhea, Dementia (eventually Death, if untreated). |
|
|
NORMAL PROTEIN DEGRADATION (TURNOVER)
|
Normal physiological process: replaces old/misfolded proteins
Proteins tagged for degradation by ubiquitin molecules (ATP-dependent) Ubiquitinated proteins transported to cytosolic proteasome where they are cleaved into peptide fragments Further degraded by non-specific proteases. AA and ubiquitin molecules are recycled. In a healthy adult, AA are incorporated into new proteins at a rate equal to the rate of protein degradation. |
|
|
NON-ESSENTIAL AMINO ACID (NEAA) BIOSYNTHESIS
|

Most of the NEAA are formed from various precursors (intermediates of glycolysis and TCA cycle)
There are only two exceptions: Tyrosine (Y) is formed from Phenylalanine (F) Cysteine is derived from serine and homocysteine, which is a product of methionine metabolism). |
|
|
Transamination Reactions
|
Reaction occurs in all cells, requires no ATP, and is readily reversible
Used for direct synthesis of three of the nonessential AA: Pyruvate (from glycolysis) → Alanine Oxaloacetate (from citric acid cycle) → Aspartate α-ketoglutarate (from citric acid cycle) → glutamate (important for disposal of ammonia) B6-mediated transfer of nitrogen group from one compound to another. |
|
|
diverse end products of AA
|
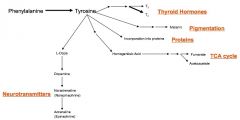
|
|
|
Phenylketonuria (PKU)
|
Defect in Phenylalanine hydroxylase (PAH) or in essential cofactors (e.g. Tetrahydrobiopterin) inhibits conversion of Phenylalanine → Tyrosine.
Elevated phenylalanine in blood and tissues Excess phenylalanine converted to phenylketones (excreted in urine) Tyrosine becomes essential; end products of phenylalanine metabolism can still be made if tyrosine obtained in diet! Mental retardation, failure to walk/talk, hypopigmentation, etc. Requires immediate and lifelong restriction of phenylalanine plus supplementation of tyrosine in the diet |
|
|
Albinism
|
Defect in Tyrosine metabolism results in deficiency of melanin production.
Multiple modes of inheritance due to several different genes involved in melanin synthesis. Complete Albinism: (Tyrosinase negative oculocutaneous albinism) results from a deficiency of tyrosinase activity, causing total absence of pigment as well as vision defects and photophobia |
|
|
Alkaptonuria
|
Deficiency in homogentisic acid oxidase causes accumulation of homogentisic acid (breakdown product of Tyrosine).
Results in homogentisic aciduria (urine turns dark upon standing), large joint arthritis, black onchronotic pigmentation of cartilage/collagenous tissue Usually asymptomatic until age 40. Low protein diets (especially low in F and Y AA) can help |
|
|
METHIONINE METABOLISM
|
Methionine is an essential AA in diet.
First residue in all protein synthesis encoded by start codon (AUG) often removed post-translationally from N-terminal end of protein (and recycled) Used in synthesis of S-adenosylmethionine (SAM) involved in addition of carbons to biological substrates Can be used to make cysteine Methionine → S-adenosylmethionine (SAM) → homocysteine Homocysteine + Serine → → cysteine *With Vitamin B6, homocysteine can condense with serine to form cysteine. |
|
|
How do you get rid of a toxin?
|
By modifying it. Can use normal metabolic pathways in body.
By medicating with B6, B12, and folic acid (folate), can force regeneration of methionine and SAM production, which allows methylation of toxins (by SAM). |
|
|
Homocystinuria
|
Most commonly caused by a defect in cystathionine synthase
Leads to high levels of methionine and homocysteine in the plasma and urine; low levels of cysteine. Manifests as ectopia lentis, skeletal abnormalities, osteoporosis, mental retardation, and atherosclerosis. |
|
|
Pernicious Anemia
|
Deficiency of vitamin B12 resulting in excess homocysteine and deficiency in THF (folate) due to the accumulation of THF in 5’-methylated form.
THF is needed in synthesis of thymine nucleotide from uracil (dUMP → dTMP). Can’t make DNA at normal rate, which results in anemia (because RBCs are rapidly turned over). Commonly observed in older white males of Scandinavian origin |
|
|
folate, cysteine, SAM, B6 and B12
|
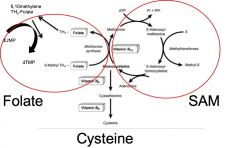
|
|
|
Histamine
|
Vasodilator that is released by degranulating mast cells
Mediates allergic and inflammatory reactions (plays other roles as well) Synthesized from histidine via pyridoxal phosphate-dependent decarboxylation reaction NB: pyridoxal phosphate is the active form of vitamin B6 (recall transamination reactions from last lecture) |
|
|
Serotonin
|
Found in intestines, brain, and platelets
Many functions: pain, sleep, body temp, blood pressure Synthesized from tryptophan via pyridoxal phosphate-dependent decarboxylation reaction |
|
|
Creatine
|
Synthesized from arginine and glycine via methylation by SAM
Phosphorylated creatine = Phosphocreatine Donate inorganic phosphate group to ADP to regenerate ATP Rapidly mobilized storehouse of energy for muscle Total body phosphocreatine is proportional to muscle mass (hard to measure) Phosphocreatine degrades to creatinine, cleared by kidney. High serum creatinine indicates failing kidney Low urine creatinine reflects decreased total muscle mass |
|
|
amino acid anabolism
|
AA come from diet, protein turnover, and de novo synthesis
Genetic errors in these pathways have clinical consequences due to downstream deficiencies or accumulation of precursors upstream |
|
|
Catabolism Preview
|
Some AA are broken down to form other AA or special products.
Carbon skeletons of AA can be broken down to provide energy by entering intermediates into gluconeogenesis and/or ketogenesis |
|
|
AMINO ACID METABOLISM: CONSUMPTION FOR ENERGY
Nitrogen |
First step in breaking down AA: take off nitrogen (toxic to CNS)
In liver (and only in the liver) make urea (non-toxic) Urea has two nitrogens, very small/soluble; only tissue through which it can’t defuse is bladder (so nitrogen can be easily excreted) |
|
|
AMINO ACID METABOLISM: CONSUMPTION FOR ENERGY
AA Carbon Skeleton Fates |
Ketogenic AA
Converted to Acetoacetyl-CoA or Acetyl-CoA Generate ketone bodies Gluconeogenic AA Can never regenerate glucose via Citric Acid Cycle (acetyl-CoA) because carbons are lost as CO2. AA recover glucose via two key pathways: 1.Pyruvate → Oxaloacetate via mitochondrial pyruvate carboxylase (requires ATP and CO2) 2.Oxaloacetate → PEP → Glucose via cytoplasmic enzyme phosphoenolpyruvate carboxykinase (requires GTP) |
|
|
AMINO ACID METABOLISM: CONSUMPTION FOR ENERGY
|
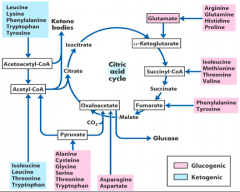
|
|
|
BRANCHED CHAIN AMINO ACIDS (BCAA)
|
Not metabolized in liver (site of most other AA metabolism)
Instead metabolized in peripheral tissues; primarily muscle 3 (essential) AA: isoleucine, valine, and leucine |
|
|
Maple Syrup Urine Disease (MSUD)
|
Deficiency in branched-chain ketoacid dehydrogenase
Autosomal recessive (often heterozygous) affects newborns within first few days of life. Accumulation of BCAA and corresponding α-keto acids interferes with brain function Metabolic acidosis (severe) Feeding problems, vomiting, dehydration Maple syrup odor to urine Mental retardation, physical disabilities, death (if untreated) |
|
|
forms of MSUD
|
Different Forms: different levels of severity of the disease depending on where the mutation occurs along the gene.
Classic Form: little-to-no enzyme activity, onset within first few days, lethal unless treated with dietary formula low in BCAA. Intermediate Form: some enzymatic activity, milder symptoms, onset between infancy and adulthood. Rare Thiamine-Dependent MSUD: most interesting form (interesting because you can do something about it); can increase enzymatic activity with bolus of thiamine. |
|
|
TOXIC AMINO GROUPS
|
Free amino groups (byproduct of AA breakdown) are toxic to the CNS because deplete TCA cycle intermediate and reduce ATP.
They must be transported safely to the liver which can process them Cannot transport as free nitrogen! 2x systems for transport (depending on where the N is coming from). Both involve transamination reaction: Peripheral tissues: N + glutamate → Glutamine (carrier molecule). Catalyzed by glutamine synthetase (requires ATP) Muscles: N + pyruvate → Alanine (carrier molecule). Catalyzed by alanine amino-transferase |
|
|
FATES OF TOXIC AMINO GROUPS
|
In the liver, reconvert Glutamine → Glutamate and Alanine → pyruvate
Glutamine donates its first N for urea synthesis when it is converted to Glutamate (catalyzed by glutaminase) Glutamate donates its N for urea synthesis in two ways |
|
|
Glutamate donates its N for urea synthesis in two ways
|
Deamination: releases NH3 directly to mitochondria (in the liver) -- can be dangerous (toxic to cells) and so the liver only wants to release N when you can capture/process it right away. Catalyzed by glutamate dehydrogenase:
glutamate → α-ketoglutarate + NH3 Transfer of amino group to oxaloacetate to form aspartate in cytoplasm (which then enters the urea cycle) catalyzed by aspartate aminotransferase (AST): oxaloacetate + glutamate → α-ketoglutarate + aspartate |
|
|
Alanine “drops” its N in liver via rxn
|
alanine + α-ketoglutarate → glutamate + pyruvate
pyruvate can return to systemic circulation for use in CA cylce |
|
|
nitrogen disposal image
|
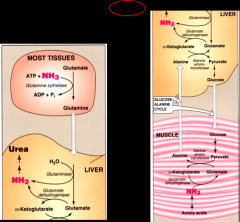
|
|
|
Transamination Reactions Involved in Transfer/Disposal of AA
|
Alanine aminotransferase (ALT)
In muscle: converts pyruvate → alanine In liver: converts alanine → pyruvate High levels of ALT in serum indicates damage to liver cells Aspartate aminotransferase (AST) Transfers amino group from glutamate to aspartate High levels of AST in the serum indicate damage to liver cells |
|
|
Glutamate Dehydrogenase
|
Unique: the only enzyme in the body that can remove nitrogen from a compound like glutamate and put it into α-ketoacid form
In Kidney: NH3 removed and excreted directly In Liver: reversible glutamate ←→ α-ketoglutarate in mitochondria of liver: rxn generates α-ketoglutarate and NH3 (for disposal of NH3 via urea cycle) in cytoplasm of liver: rxn regenerates glutamate from alanine and α-ketoglutarate Requires NAD and NADP (does not require ATP) Can be coupled with transamination to remove N from excess AAs |
|
|
What do we do with the Nitrogen?
|
Put it into urea.
Urea is only synthesized in hepatocytes of the liver (Kidney excretes NH3 directly) |
|
|
Urea Cycle
|
Initial step (rate limiting): carbamoyl phosphate synthetase I generates carbamoyl phosphate from NH3, CO2, and 2ATP (in mitochondria of liver)
NH3 comes from glutamate or glutamine carbamoyl phosphate → citrulline (shuttled to cytosol of liver) citrulline + aspartate → argininosuccinate → fumarate + arginine Final step: arginine → ornithine + urea final step catalyzed by Arginase: key urea cycle enzyme that is present ONLY in the liver. |
|
|
where is the urea cycle taking place?
|
Two steps occur in liver mitochondria: (1) formation of carbamoyl phosphate and (2) adding of carbamoyl phosphate onto ornithine. Everything else occurs in the cytoplasm.
|
|
|
urea cycle
|
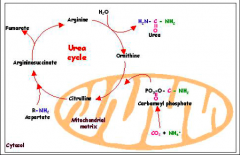
|
|
|
Hyperammonemia
|
Excess free ammonia in body (i.e. ammonia not removed via urea cycle)
Halts energy production in the cells of the brain (by removing a key TCA cycle intermediate) Can be: *Genetic - most commonly ornithine transcarbamylase deficiency *Acquired - most commonly caused by liver disease Signs of toxicity: tremors, slurred speech, somnolence, vomiting, cerebral edema, blurring of vision Prescription: limit protein in diet; administration of drugs that bind ammonia and promote safe excretion *Phenylbutyrate -- prodrug that is rapidly converted to phenylacetate, which combines with glutamine and is excreted in urine |