Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
237 Cards in this Set
- Front
- Back
|
What’s the halflife of RBCs/granulocytes/platelets? Of Transfused cells?
|
Erthrocytes have a total life span of 120 days, granulocytes about 10 hours in the circulating blood, and platelets 10 days.
Transfused cells are a mixture of young and old cells, and thus their mean life span is about 28 days. |
|
|
How is anemia defined?
|
Anemia is defined as a decrease in circulating hemoglobin (Hb) concentration. Normal values—men: 15.5 g/dL, women: 13 g/dL (pregnancy: 11 g/dL).
|
|
|
How many molecules of oxygen can Hb bind?
|
Hb can bind 4 molecules of oxygen, because it has 4 heme units, and each heme group can bind one oxygen molecule.
|
|
|
What cells make platelets, and how do platelets enter the circulation?
|
Megakaryocytes make platetlets. They mature in the bone marrow sinuses in osteoblastic niches. They then become adherent to vessels and protrude cytoplasmic proceses (proplatelets) through endothelial gaps into the vascular lumen. These detach and circulate as platelets.
(proplatelets) |
|
|
What is the red cell volume in an 70 kg person?
|
2.2 L. Men: 32 mL/kg Women: 26 mL/kg.
|
|
|
How do you define the MCV, MCH, and MCHC? RDW?
|
Mean Corpuscular Volume (MCV): Hct/RBC – The average volume of a red cell
Mean Corpuscular Hemoglobin (MCH): Hb/RBC – The average content of Hb per red cell Mean Corpuscular Hemoglobin Concentration (MCHC): Hb/Hct - The average concentration of Hb in a given volume of packed red cells Red Cell Distribution Width (RDW): Standard deviation of MCV/MCV = 11.5-14.5% (extent of variability in RBC size) |
|
|
Describe the breakdown of heme from RBCs?
|

When RBCs are degraded, the heme is detached from the globin, and the enzyme heme oxidase converts heme to biliverdin+CO+iron. Biliverdin is reduced to bilirubin for excretion in the bile. Bilirubin, bound to albumin (indirect bilirubin) is transported to the liver where glucuronide is attached (direct bilirubin) prior to excretion in the bile. In the gut, bilirubin is converted to urobilinogen and either excreted as fecal urobilinogen or reabsorbed and excreted by the kidney as urine urobilinogen. The iron that was attached to the heme is liberated and re-utilized.
|
|
|
When does hemoglobin switch take place, and what is it?
|
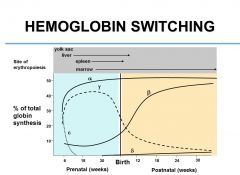
Hemoglobin switch is the transition from fetal subunits (Hb-gamma) to adult Hb subunits (Hb-beta). Hb alpha is present in both the fetus and the adult. This switch occurs at around 6-9 months after birth.
|
|
|
What intermediaries are involved in oxygen sensing, and where does this occur?
|
The juxta-tubular cells of the kidney sense oxygen tension. Proline hydroxylase requires O2 to hydroxylate a key protein: hypoxia-inducible factor (HIF). This allows the ubiquitination of HIF-alpha by the von Hippel-Lindau factor (VHL) & ubiquitin ligase (UL), resulting in the degradation of HIF in the proteasome. When O2 is low, HIF activates the gene for EPO and other genes, increasing Hb and encouraging blood vessel formation.
|
|
|
Which way does the Hb-O2 dissociation curve shift in the presence of anemia or acidosis?
|
The Hb-O2 dissociation curve shifts right when there’s anemia, acidosis, or plentiful 2,3 DPG so that at a given PO2, the Hb O2 saturation is less, and more O2 is delivered to peripheral tissues.
|
|
|
What volume of RBCs is produced daily?
|
18 mL of RBC produced daily. Since RBC lifespan is 120 days (2200 mL/120 days = 18 mL/day to maintain steady state).
|
|
|
How much is daily iron absorption?
|
1-2 mg iron absorbed daily in the iron replete person.
|
|
|
When is transferrin increased? Decreased?
|
Transferrin levels are increased in…
Iron Deficiencies Transferrin levels are decreased in… Gene defects (very rare) liver disease, malnutrition (Impaired synthesis) renal or bowel disease (Loss in urine or stool) Inflammatory disorders (binding to macrophages) |
|
|
What is hepcidin, and what is its role in iron metabolism?
|
Hepcidin is a peptide hormone synthesized by the liver in response to iron overload and inflammatory cytokines such as IL-6. Hepcidin downregulates ferroportin, leading to decreased iron absorption and decreased iron release from macrophages.
Iron deficiency and hypoxia decrease hepcidin production, resulting in increased iron absorption and greater iron availability to erythroblasts and other parenchymal cells. Dysregulation of hepcidin, as in hemochromatosis, results in iron overload. |
|
|
How much iron is needed to produce 1 mL of red cells?
|
1 mL of red cells contains 1 mg of iron, and 1 mg of iron is needed to produce 1 mL of red cells.
|
|
|
What disorder should be considered if all RBC indices (MCV, MCH, MCHC) are low?
|
If all RBC indices are low, consider iron deficiency or thalassemia. RBCs are small & hypochromic due to impaired Hb synthesis.
|
|
|
What disorders should be considered in the case of megaloblastic anemia?
|
Consider folate or B12 deficiency with high MCV (megaloblastic RBCs), because RBC maturation is impaired.
|
|
|
What disorder(s) should be considered if MCHC is high?
|
If MCHC is high, consider spherocytic anemia, because more Hb can be packed into a sphere than a biconcave disk.
|
|
|
When is the Red Cell Distribution Width (RDW) high?
|
RDW is high in iron deficiency (small and nl RBC) and B12 and folate deficiency (nl and large RBCs). Otherwise it’s normal.
|
|
|
Where do you expect to find RBC production in the adult?
|
By the age 20, production marrow becomes limited to proximal femurs, pelvic bones, vertebrae, ribs, and skull.
|
|
|
What is the only type of totipotent cell?
|
A fertilized egg is the only example of a totipotent cell, capable of unlimited capacity to give rise to any cell type.
|
|
|
What is pluripotency?
|
Pluripotency is the ability to give rise to most tissues of an organism.
|
|
|
What is multipotency?
|
Multipotent stem cells are specialized stem cells that are committed to give rise to a cell with a specific function.
|
|
|
If an elderly gentlemen presents with decreased RBCs and fatigue, what should you suspect?
|
You should suspect iron deficiency anemia due to bleeding. The bleeding is most likely due to cancer of the GI tract.
|
|
|
How much iron is absorbed on a daily basis versus what is consumed?
|
Out of an average of about 15 mg of Fe consumed daily, only 1-2 mg is absorbed.
|
|
|
What is the limiting factor in your ability to make blood?
|
Fe is the limiting factor in your ability to make blood.
|
|
|
How much blood is in a unit of blood?
|
500 mL is in one unit of blood. 45% of that is red cells.
|
|
|
What is the most common cause of Fe deficiency in a child? In an adult? What is the most common cause of the top cause of Fe deficiency in the adult?
|
Child: malnutrition
Adult: Bleeding, most commonly due to hemorrhoids |
|
|
What are physical exam findings consistent with Fe-deficiency anemia (IDA)?
|
Physical exam: bluish sclera, smooth tongue, fissured nails
|
|
|
What are most common symptoms of hemochromatosis?
|
1) Fatigue
2) Hyperpigmentation 3) Arthritis |
|
|
What are the 3 different classes of genetic hemochromatosis disorders?
|
Class I affects hepcidin itself (mutation in HFE decreases hepcidin production)
Class II (most common type) affects hepcidin regulation via HFE, TFR2, or Hemujuvelin (HFE2) Class III affects ferroportin and thus induces hepcidin resistance |
|
|
What are the dangers of “free” iron?
|
Free iron can generate hydroxyl radicals which can cause organelle damage or fibrosis.
|
|
|
How is hemochromatosis treated?
|
Hemochromatosis is treated with phlebotomy. The goal is to decrease transferrin saturation so that iron will transfer from tissues to blood.
|
|
|
How much iron is needed on a day-to-day basis for homeostasis?
|
20 mg; 20 mL of RBCs are recycled daily for Fe.
|
|
|
What is the first pathogenic mechanism in anemia of inflammation?
|
The first pathogenic mechanism in AI involves direct suppression of erythropoiesis through erythroid progenitor suppression by pro-inflammatory cytokines under inflammatory conditions (esp. IL-1, TNF, and IFN). IL-1 requires IFN-gamma, and TNF requires IFN-beta.
|
|
|
Besides direct suppression of erythropoiesis, how does inflammation induce anemia?
|
* Inflammation upregulates hepcidin (esp. cytokine IL-6 through the JAK-STAT pathway)
* inflammation blunts EPO expression, particularly with IL-1 and TNF. * AI shortens RBC survival (not elaborated in lecture) |
|
|
What molecules regulate hepcidin upregulation/suppression?
|
Hepcidin suppression is mediated by TMPRSS6, while hepcidin is upregulated by BMP-6/SMAD signaling, including accessory proteins like HFE, Hemojuvelin (HJV), and Transferrin Receptor 2.
|
|
|
Why would it not be effective to give iron to someone with anemia of inflammation or inflammation in general with IDA?
|
AI is a disease of iron sequestration. They have a lot of iron stored, but it’s not available for use due to the upregulation of hepcidin (caused by inflammation via IL-6), which disables the flow of iron via ferroportin from enterocytes to the circulation.
|
|
|
What diagnostic markers would differentiate IDA from AI?
|
* Both IDA and AI have low serum iron and low transferrin saturation
* AI has high/increased soluble transferrin receptor * In AI, you would have increased ferritin due to Fe sequestration caused by upregulated hepcidin, whereas IDA has low ferritin * In AI, transferrin would be low, whereas it’s high in IDA * In AI, transferrin receptor is low, whereas it’s high in IDA * In AI, EPO is not appropriately increased, whereas it’s high in IDA * AI would also have increased inflammatory markers as support (ESR and CRP) |
|
|
How do you treat AI?
|
• Treat the underlying disease
• Give Fe only if the patient is deficient |
|
|
What is the most common inherited RBC membrane disorder, and what is its corresponding pathogenic mechanism?
|
Hereditary spherocytosis is the most common inherited RBC membrane disorder, affecting up to 1 in 3000 individuals of northern European descent. It’s autosomal dominant in 75% of cases and most commonly caused by ankyrin deficiency (followed by spectrin and less commonly band 3 deficiency).
|
|
|
What happens to a RBC if you expand the outer layer without changing the inner layer? What about if you expand the inner layer without expanding the outer layer?
|
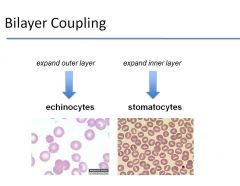
1) If you extend the outer layer without changing the inner layer -> echinocytes
2) If you extend the inner layer without expanding the outer layer -> stomatocytes |
|
|
What happens to RBCs in instances of loss of surface area without change in volume? What about a gain in volume without a change in surface area? What about surface gain? Volume loss?
|
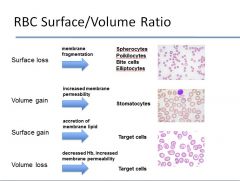
see image
|
|
|
What protein loss results in Hereditary Elliptocytosis? Southeast Asian Ovalocytosis?
|
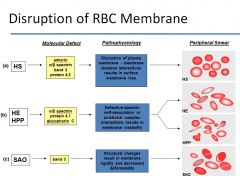
see image
|
|
|
What does glucose-6-phosphate dehydrogenase do?
|
G6PD plays a major role in converting glucose-6-phosphate to generate NADPH equivalents. NADPH reduces glutathione which in turn protects the RBC from oxidative stress.
|
|
|
What is the inheritance pattern of G6PD deficiency?
|
G6PD is an X-linked inherited disorder, so it will mostly affect males, although females carriers could be affected through lyonization.
|
|
|
What is pyruvate kinase deficiency, and how does it lead to disease?
|
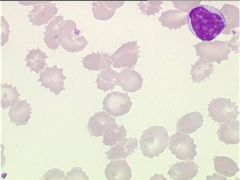
Pyruvate kinase deficiency is the most common cause of non-spherocytic hemolytic anemia due to defective glycolysis in the Caucasian population (pyruvate kinase catalyzes the major, last step in ATP formation). PK deficiency causes 2 major metabolic derangements:
1) ATP depletion 2) Increase in 2,3 DPG production (this is why they appear normal and are not tired at all—better oxygen delivery) It’s inherited in an autosomal recessive manner and results in variable anemia, jaundice and splenomegaly. PK deficiency produces RBCs with thorny projections (echinocytes). |
|
|
Why does parvovirus cause aplastic crisis in hemolytic disorders?
|
Parvovirus causes aplastic crises in hemolytic disorders, because it goes to the bone marrow and specifically shuts down RBC production.
|
|
|
Why is MCHC elevated in hereditary spherocytosis (HS)?
|
The decrease in surface area causes more Hb to be packed in a smaller area, thus increasing the MCHC. Elevated MCHC is a very sensitive and specific clue that HS is the underlying cause of disease.
|
|
|
What are diagnostic clues to cue you to hereditary spherocytosis (HS)?
|
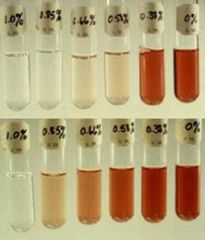
o Peripheral smear
o Laboratory features • Variable anemia • Reticulocytosis • Elevated MCHC – Fairly sensitive + specific • Elevated total bilirubin o Osmotic fragility test • HS pts will demonstrate high hemolysis at almost any concentration of NaCl • A normal RBC, since it’s biconcave, can expand and accommodate extra influx of H2O, whereas a spherical RBC cannot |
|
|
What causes Southeast Asian Ovalocytosis?
|
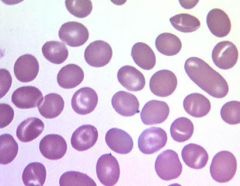
o Autosomal dominant
o Caused by band 3 deficiency o Mild anemia despite marked RBC rigidity. Not a lot of clinical problems |
|
|
Why should people with G6PD deficiency avoid fava beans?
|
Fava beans present a large oxidative load, which those with G6PD deficiency cannot handle due to reduced NADPH, which handles ROS via glutathione.
|
|
|
What is the reason for the formation of gallstones in Hereditary Spherocytosis (HS)?
|
The hemolysis of RBCs leads to an increase in bilirubin which can cause the formation of gallstones.
|
|
|
What are the three types of situations in which drugs could cause hemolytic anemia?
|
1) The drug could bind to RBCs, and antibodies could be formed against the drug, eventually causing RBC hemolysis – penicillin & cephalosporins
2) Ab can bind both the drug (which is bound to RBCs) and the RBC at the same time - quinine 3) Drug stimulates the production of an auto-antibody directly to RBCs (not known why this occurs) – alpha methyldopa, levodopa, procainamide |
|
|
In what type of hemolysis would you see a reading that is positive for complement in a DAT?
|
Cold Agglutinin Disease (cold antibody hemolytic anemia)
|
|
|
What test would you perform to detect the presence of allo-antibodies?
|
Indirect Coombs test.
|
|
|
What is the pathogenesis of Spur Cell Anemia?
|
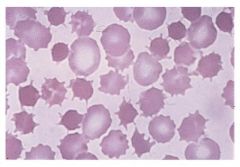
Spur cell anemia occurs in chronic, severe liver cirrhosis where patients have excess cholesterol which attaches to RBCs and causes membrane defects. It can be treated with splenectomy, but the best treatment would be liver transplant.
|
|
|
What is the pathogenesis of paroxysmal nocturnal hemoglobinuria (PNH)?
|
There are mutations in GPI-A which can cause GPI (Glycosylphosphatidylinositol)-anchored protein deficiencies, which leads to complement-mediated lysis of cells. You can have acute hemolysis overnight, leading to blood in the urine when they wake up.
|
|
|
How is the Schilling test performed, and what is it used for?
|
The Schilling test is used to confirm vitamin B12 deficiency.
1st part A patient is given an oral dose of radiolabeled B12 and an intramuscular injection to saturate the serum/liver binding sites in a nuclear medicine lab. If radiolabeled B12 is absorbed, it will be excreted in the urine (high radioactivity in urine). However, if the urine radioactivity count is low, then you know that malabsorption is present. 2nd part All steps same as part one except use intrinsic factor concurrently with radioactive vit B12. Individuals with pernicious anemia will have high urine levels of radioactivity, whereas those with malabsorption due to a separate cause will have low radioactivity in the urine. |
|
|
How would megaloblastic anemia affect the bone marrow and blood counts?
|
In megaloblastic anemia, you would see hypercellular bone marrow packed with precursor cells.
You would also expect anemia, thrombocytopenia and leucopenia as a result of impaired DNA synthesis (combination of hypercellularity+low blood count = ineffective erythropoiesis). |
|
|
What cellular morphological features are hallmarks of megaloblastic anemia?
|
In megaloblastic anemia, you have an increased MCV due to large precursor redblood cells, like macrocytes and macro-ovalocytes.
You would also expect to see hypersegmented neutrophils in megaloblastic anemias. |
|
|
How would homocysteine levels change in vitamin B12 or folate deficiency and why?
|
Homocysteine levels rise in vitamin B12 or folate deficiency, because they both participate in converting it to methionine.
|
|
|
How do the body stores of folic acid differ from that of vit B12 deficiency?
|
Vit B12: 2-4 years
Folic acid: 3-4 months |
|
|
What specific neuropathy is characteristic in pernicious anemia?
|
Loss of vibratory sense and position sense are characteristic neuropathic features in pernicious anemia.
|
|
|
What two molecules are bound to heme in its reduced state?
|
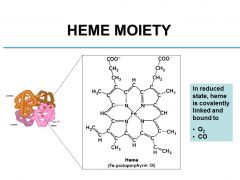
O2, CO
|
|
|
What are prenatal sites of erythropoiesis?
|
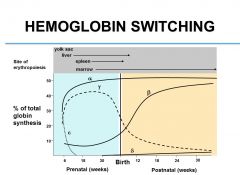
Yolk sac
Liver Spleen Marrow |
|
|
What is the normal distribution of functional hemoglobin (in terms of HbA, HbF, HbA2.)?
|

90-97% HbA
~1% HbF ~2% HbA2 |
|
|
What factors shift the O2-Hb dissociation curve to the right?
|
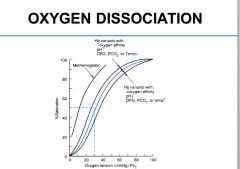
Decreased pH, Increased DPG, PCO2 or temperature
|
|
|
What genetic mutation produces SCD?
|
A single nucleotide substitution in the 6th codon of the beta gene produces the mutation in the beta globin chain. GAG (glutamate) ->GTG (valine)
|
|
|
What is the primary event in the pathogenesis of SCD?
|
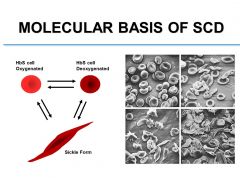
Intracellular polymerization of HbS during deoxygenation is the primary event in the pathogenesis of SCD. Highly ordered rigid helical polymers stretch the blood cell and give it a sickle shape. Polymerization occurs only in deoxygenated state.
|
|
|
Where is the most sensitive site to sickling?
|
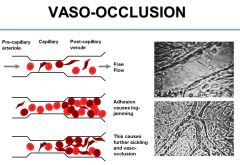
Most problems occur in post capillary venules, because O2 is lowest on that side. HbS cells tend to sickle, abnormal stage damages vascular endothelium and increases expression of various molecules. They stick to surface of blood vessel in that area. Log jamming effect creates hostile extracellular environment, because you’re decreasing the pH, which can cause further sickling
Eventually that dissipates. Oxidative stress can occur. |
|
|
What factors/mechanisms contribute to vaso-occlusion?
|
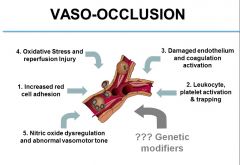
see image
|
|
|
Why are newborns with sickle cell disease not usually anemic?
|
Newborns with sickle cell disease make HbF which is protective.
|
|
|
What is most common cause of death with sickle cell disease?
|
Infection represents most common cause of death. Functional asplenia is the biggest risk factor, as the spleen is important for the immune response against encapsulated organisms (e.g., Streptococus pneumonia, Haemophilus influenza, Neisseria meningitides).
|
|
|
What is Acute Chest Syndrome (ACS)?
|
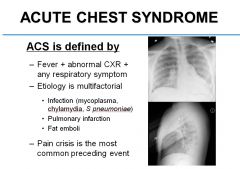
ACS is defined by Fever, abnormal CXR, and any respiratory symptom. Its etiology is multifactorial and could be due to infection, pulmonary infarction, or fat emboli.
|
|
|
What is the single most important factor in decreasing morbidity due to SCD?
|
Newborn screening.
|
|
|
What is the only FDA approved medicine to treat SCD?
|
Hydroxyurea; it reduces hospitalizations/ER visits, ACS, pain, and increases the expression of HbF.
|
|
|
What does “balanced polymorphism” mean?
|
The heterozygous state has a protective effect, but the homozygous state is associated with premature death due to disease.
|
|
|
What causes the classic “sickle form” of RBCs in SCD (i.e., what is the pathogenic mechanism of sickling)?
|
The sickle form is due to decreased solubility of Hb S under hypoxic conditions and increased polymerization. The deoxygenated Hb S polymers form highly ordered fiber aggregates, elongate the cell, and distort it.
|
|
|
What is the Gardos channel, and how does it relate to sickling?
|
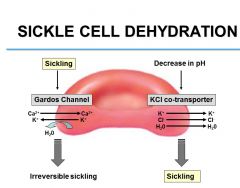
Sickling activates Gardos channel and promotes efflux of water and leads to mostly reversible but some irreversible sickling
The more sickling, the more the cells get stuck in the microcirculation, and the extracellular environment becomes very hostile, the greater the pH decreases (more acidic environment), and the excess H+ ions enter red cell and activate the KCl cotransporter, and there’s an efflux of KCl and water and furthers sickling |
|
|
What is the operational definition of Thalassemia Major?
|
Thalassemia major is a transfusion-dependent anemia often due to homozygous beta-0 mutations or compound heterozygous for beta-0 and beta-+ mutations. Operational definition: Require 8 or more transfusions/year.
Intermedia receive less than 8 transfusions/year. Minor (thal trait): Heterozygous Beta-globin gene mutation. |
|
|
Describe the different disease processes as you accumulate mutations/deletions in the 4 different alpha Hb genes.
|
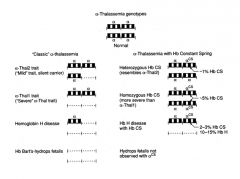
see image
|
|
|
What is HbE disease?
|
• Occurs in 30-45% of some Southeast Asian populations
• Results from a single amino acid substitution caused by a point mutation in the ß globin gene • Homozygous Hb E is associated with a relatively benign clinical course • Coinheritance of Hb E with ß thalassemia results in a condition similar to ß thalassemia major in severity |
|
|
What is HbH disease?
|
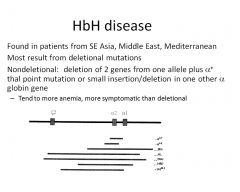
see slide
|
|
|
What is Hb Constant Spring?
|

|
|
|
What is Hb Bart?
|
HbBart is gamma-tetramers formed due to excess gamma-globin in fetal development (lack of functional alpha in alpha thalassemia).
|
|
|
What tetramers are most common in alpha thalassemia?
|
HbH (Beta globin chain tetramers)
|
|
|
Describe the pathophysiology of Beta thalassemia?
|
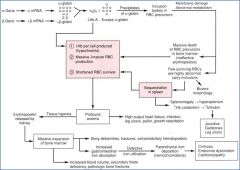
see image
|
|
|
What tools could you use to diagnose thalassemia?
|
• High Performance Liquid Chromatography
o Allows for efficient reproducible separation of normal and common abnormal globin variants o Method used by state of Illinois newborn screening program • Hemoglobin Electropheresis o Separation of normal and most common abnormal globin variants o Performed at both alkaline and acid pH o Cannot quantify HgbA2 o Test used for confirmatory testing for beta globin defects • DNA analysis • Complete Blood Count |
|
|
How does diagnosis for thalassemia differ in the newborn versus adults?
|

Neonatal: 1) HPLC 2) Electrophoresis
Adult: 1) CBC 2) Hb electrophoresis |
|
|
Which chromosome is the beta globin gene on? The alpha globin gene?
|
The beta globin locus is on chromosome 11, and the alpha is on 16.
|
|
|
If you have a 9 month old baby present with microcytic, hypochromic anemia with increased RBCs, what is the likely diagnosis?
|
Thalassemia
|
|
|
What would you expect to see in the peripheral blood smear in a 2 month old with alpha thalassemia? Beta thalassemia?
|
In alpha thalassemia, before 3 months you can catch HbBart’s. In Beta thalassemia, the baby would have HbF only.
|
|
|
What RBC disorder would you expect in a 1 year old with increased HbF and increased HbA2?
|
Beta-Thalassemia
|
|
|
What are ways to measure/monitor iron overload, and what are their benefits/drawbacks?
|
Serum ferritin concentration is a simple, noninvasive way to measure iron overload, but it has an imprecise correlation with the body iron.
Tissue iron is a better measure of iron overload, but it’s not simple to measure. |
|
|
What are all the common places of iron deposition in beta thalassemia patients who receive blood transfusions?
|
Heart (most clinically significant), thyroid, parathyroid, liver
|
|
|
What causes the “Hair-on-End” skull deformity in thalassemia major?
|
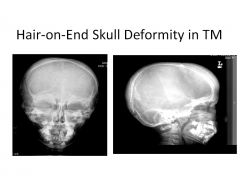
Marrow expansion causes the Hair-on-End phenotype in thalassemia major: ineffective erythropoiesis (defective RBCs die in the marrow).
|
|
|
What causes short stature in thalassemia patients?
|
Thalassemia patients have premature closure of their epiphyses. They can also have decreased amounts of GH due to pituitary changes.
|
|
|
What is the SCCS?
|
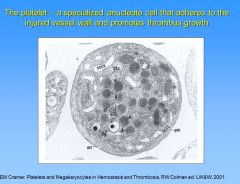
Surface cannon canalicular system (SCCS) is a redundant membrane that reaches into deep part of platelets, and upon activation, provides easy access for granules to be released. Also involved in shape change of the platelet.
|
|
|
What molecules are produced by endothelial cells that have platelet inhibiting function?
|
• NO is a platelet inhibitor
• PGI2 is a platelet inhibitor • CD39 is a receptor that’s able to take ADP and degrade it. ADP is a potent agonist of platelets, so keeping them low helps keep blood in a liquid state. |
|
|
What receptor(s) is/are expressed on the surface of activated platelets?
|
Fibrinogen receptor (GPIIb/IIIa). Fibrinogen is the most important multivalent adhesive glycoprotein that can bridge two activated platelets, but other ligands like vWF can also serve this function.
|
|
|
What molecules activate platelets when they adhere to subendothelium after vascular injury?
|
• Collagen
• Thrombin (small amounts) • vWF |
|
|
What molecules do platelets secrete to further activate more platelets?
|
• TxA2
• ADP |
|
|
What molecules are responsible for fibrinolysis?
|
• t-PA
• Plasmin (from activated plasminogen) |
|
|
What molecule is responsible for cross-linking fibrin?
|
Factor XIII allows fibrin to cross-link.
|
|
|
What is P2Y12, and what is its function in platelet activation?
|
P2Y12 is a GPCR that inhibits cAMP, which would constitutively inhibit platelet activation. Thus P2Y12 activates platelet activation by inhibiting cAMP.
|
|
|
What causes the “second wave” in the Platelet Function Analyzer assessment?
|
As platelets activate, plasma clears, and light transmittance becomes greater and greater. When platelets are activated, granules are released, and you get second wave. If platelets are inhibited, granules are not released, and the second wave will have lower amplitude.
|
|
|
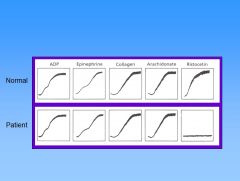
What defect does this pt have?
|
This patient has Glanzmann thrombasthenia – GP IIb/IIIa mutation. In this pt, there’s no functional fibrinogen receptor, so the platelets cant aggregate no matter what they’re stimulated with. Ristocetin is an Abx related to vancomycin (causes platelets to stick together and thus no longer used). It binds vWF and makes it sticky for platelets. Shown is normal agglutination of platelets due to ristocetin, showing vWF is normal. But the fibrinogen receptor is not working.
|
|
|
What defect does this pt have?
|
This patient has Bernard-Soulier syndrome, which is the opposite of Glanzmann thrombasthenia. The vWF receptor is defective. Multiple defects can lead to Bernard-Soulier syndrome. They get giant platelets in B-S syndrome.
|
|
|
What defect does this pt have?
|
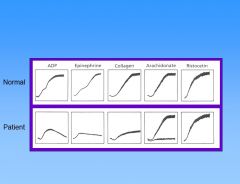
This pt has storage pool defect. Storage pool deficiency are a heterogeneous group of disorders in which the patients have deficiency of platelet dense granules (d-SPD), alpha granules (a-SPD) or both granules (ad-SPD) and manifest mild to moderate bleeding disorders. Platelet aggregometry shows primary aggregation wave with loss of secondary aggregation wave.
|
|
|
What is the mechanism of action of Aspirin?
|
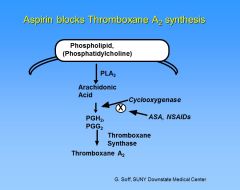
Aspirin blocks thromboxane A2 synthesis by irreversibly acetylating the cyclooxygenase enzyme. This pathway is blocked for the life of the platelet. NSAIDs also block cyclooxygenase, but the effect is reversible and returns to normal after the drug is withdrawn.
|
|
|
What is the mechanism of action and function of clopidigrel and ticlopidine?
|
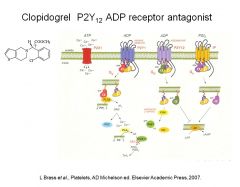
Clopidogrel and ticlopidine have antithrombotic effects by irreversibly inhibiting the ADP-induced platelet aggregation by blocking the P2Y12 receptor, which is involved in platelet activation.
|
|
|
What is the mechanism of action and function of dipyridamole?
|
Dipyridamole has antithrombotic effects by increasing platelet cAMP levels.
|
|
|
What is the mechanism of action and function of Abciximab?
|
Drugs like Abciximab, tirofiban, and eptifibatide have antithrombotic effects by inhibiting GPIIb/IIIa (fibrinogen) receptors. They are mostly used in cardiac cath labs.
|
|
|
Where is Thrombopoeitin (TPO) synthesized and stored? Where does it bind?
|

TPO is constitutively synthesized in the liver, but it’s not stored. It binds to the c-MPL receptor on megakaryocytes to stimulate platelet production.
|
|
|
What is the mechanism of rituximab, and what’s a drawback of its use?
|
Rituximab is an anti-CD20 monoclonal antibody, and although it’s safe and effective, it’s very expensive.
|
|
|
What is the mechanism of action of romiplostim?
|
Romiplostim is a peptide mimetic that binds/activates c-mpl receptor to stimulate platelet production and induce maturation of megakaryocytes, although it has no sequence homology with native TPO. It’s useful for treating ITP.
|
|
|
What is the mechanism of action of ELtrombopag?
|
Eltrombopag is a nonpeptide mimetic that promotes CD34+ differentiation to megakaryocytes and thus increases platelet production.
|
|
|
What is the classic pentad for TTP?
|
1) Fever
2) neurologic abnormalities 3) renal dysfunction 4) thrombocytopenia 5) microangiopathic hemolytic anemia – when there’s a mechanical destruction of RBCs Also look for high LDH. |
|
|
What does ADAMTS13 do?
|
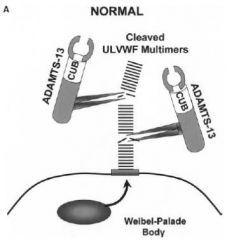
ADAMTS13 cleaves long strands of vWF and makes sure you don’t get inappropriate platelet activation.
|
|
|
What is a mechanism for idiopathic TTP?
|
In idiopathic TTP, Ab binds ADAMTS13, so it can’t cleave vWF. Platelet count goes down, because platelets are consumed, and RBCs are sheared into schistocytes. It is not known why Abs form to ADAMTS13.
|
|
|
What is the mechanism of Heparin Induced Thrombocytopenia (HIT)?
|
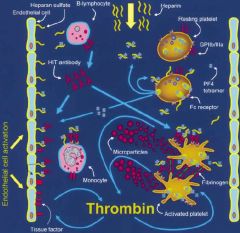
PF4-heparin-Ab binds to receptor on platelets, activates platelets (platelet levels drop), and causes a lot of thrombin to be generated. The pts are really thrombogenic and prone to develop venous and arterial blood clots. HIT occurs more often in those who receive unfractionated heparin more than molecular weight heparin.
|
|
|
When is it useful to check antibodies in the context of thrombocytopenia?
|
It’s useful to check Abs in HIT, because it’s very sensitive, unlike ITP where antibodies are neither sensitive nor specific.
|
|
|
How would you treat HIT?
|
You could treat HIT by stopping heparin and using direct thrombin inhibitors (Argatroban, lepirudin).
|
|
|
What are Weibel-Palade organelle, and what is their function?
|
Weibel-Palade organelle are structures inside endothelial cells that contain and release endothelin (vasoconstrictor), P-selectin, and vWF.
|
|
|
Why is it effective to place your finger in your mouth after puncturing it?
|
Saliva on the wound stops bleeding, because the saliva’s full of microvesicles that has tissue factor.
|
|
|
Which factor deficiency would you not see a prolonged aPTT?
|
Factor VII
|
|
|
Why don’t persons with FXII deficiency bleed? Why do hemophiliacs bleed?
|
Activated platelets contribute to thrombin generation; this does not require FXII, which explains why those with FXII deficiency do not blood.
|
|
|
What initiates clotting in vivo? In vitro?
|
In vivo, clotting is activated by tissue factor, which forms a complex with FVII. In vitro, clotting is activated by the glass test tube, which activates factor XII, initiating the intrinsic pathway.
|
|
|
How is PT tested?
|
A high concentration of tissue factor is added to form a TF-VIIa complex which activates FX; FXa with FV and calcium converts prothrombin to thrombin, and thrombin converts fibrinogen to fibrin. The clotting time becomes prolonged when the concentration of one or more clotting factors (V, VII, X, prothrombin) is less than 30% of normal.
|
|
|
How is PTT tested?
|
A silicate is added that activates FXII. A phospholipid and calcium provided accelerates the clotting time. aPTT becomes prolonged in the deficiency (<30% of normal) of pretty much any clotting factor except FVII.
|
|
|
Which factor deficiency is associated with a mild/moderate bleeding disorder?
|
FXI
|
|
|
What accounts for the incredible strength of the fibrin strand?
|
Thrombin-activated FXIII catalyzes a bond formation between the glutamic acid of one D-domain and lysine of an adjacent D-domain.
|
|
|
What anti-coagulation factors are produced by endothelium?
|
Intact endothelium produces a lot of antithrombotic agents, like:
NO, prostacyclin, ecto-ADPase, TFPI (blocks FVIIa-tissue factor complex), thrombomodulin (binds thrombin and activates protein C), |
|
|
What enzyme lyses fibrin clots?
|
Plasmin lyses fibrin clots. t-Pa and urokinase convert plasminogen to plasmin.
|
|
|
Where do microparticles come from, and what role do they play in hemostasis?
|
Microparticles come from blebs shed off leukocytes, and they have tissue factor, which activates clotting. The microparticles dock and fuse with activated platelets.
|
|
|
How is protein C activated, and what are its targets?
|
Protein C is activated by thrombomodulin, and activated protein C, with free protein S (PS), inactivates FVa and FVIIa.
|
|
|
What is the limiting factor for blood coagulation?
|
Calcium
|
|
|
What is consumptive coagulopathy, and in what circumstances does it occur?
|
Consumptive coagulopathy is a situation in which clots/microthrombi are forming and consuming the clotting factors such that the patient is at a greater risk for a bleeding problem. It occurs in hemangiomas (e.g., Kasabach-Merritt Syndrome where large hemangiomas form).
|
|
|
In what situations would you have both PT and PTT prolonged?
|
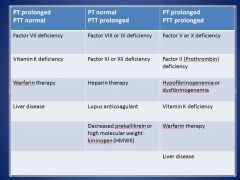
• Factor II, V, or X deficiency
• Hypofibrinogenemia/dysfibrinogenemia • Vitamin K deficiency • Warfarin therapy • Liver disease |
|
|
In what situation would PT be prolonged, but PTT normal?
|
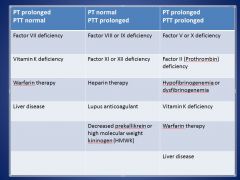
• Factor VII deficiency
• Vitamin K deficiency • Warfarin therapy • Liver disease |
|
|
In what situation would PTT be prolonged, but PT normal?
|
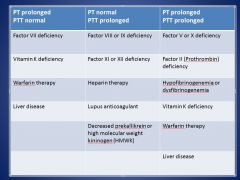
• Factor VIII, IX, XI, or XII deficiency
• Heparin therapy • Lupus anticoagulant • Decreased prekallikrein or high molecular weight kininogen (HMWK) |
|
|
Which intrinsic pathway deficiencies cause bleeding?
|
Factor VIII, IX, XI, vWF
|
|
|
Which intrinsic pathway deficiencies do not cause bleeding?
|
Factor XII, pre-kallikrein, high molecular weight kininogen (HMWK)
|
|
|
What is Hereditary Hemorrhagic Telangiectasia?
|
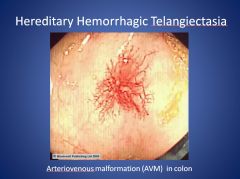
Hereditary Hemorrhagic Telangiectasia is a genetic disorder (autosomal dominant—Endoglin or ALK1 gene) that leads to abnormal blood vessel formation in the skin, mucous membranes, and often in organs like the lungs, liver, brain, and GI tract. Arterial Venous Malformations (AVMs) occur, and they may lead to nosebleeds. Diagnosis is based on clinical manifestations.
|
|
|
What is a mixing study, and what is it used for?
|
A mixing study is used in situations where you suspect a missing clotting factor due to an elevated PT or PTT (or both). You take the patient’s sample and mix it with normal plasma 1:1. If the clotting time corrects to normal, then you have evidence there’s a factor missing. If it does not correct to normal, you may have a factor inhibitor present in the plasma inhibiting even normal plasma (e.g., Lupus anticoagulant).
|
|
|
How low does the missing clotting factor have to be in order to get an abnormal PT or PTT?
|
<30-40%
|
|
|
What factor deficiency produces hemophilia A? Hemophilia B? What is their genetic inheritance pattern?
|
• Hemophilia A: VIII, Hemophilia B: IX
• They are X-linked recessive • Inversion of intron 22 in factor VIII accounts for 40% of severe Hemophilia A cases |
|
|
What are the most common sites of bleeding for hemophilia patients?
|
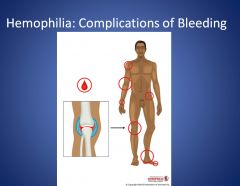
Hemophilia patients most commonly bleed in their joints. They can present with pain/stiffness in their joints (morning stiffness), swollen joints. They end up with joint damage. Swelling doesn’t go down, because there’s new bone formation there. They can end up needing a hip replacement.
|
|
|
Why are factors VIII and IX important in hemophilia?
|
Factors VIII and iX, when activated in the presence of phospholipid and calcium, form the tenase complex which activates factor X, a critical step in coagulation. Although factor X may be activated by tissue factor-factor VIIa complex, only small amounts are formed, and these are quickly inactivated by the tissue factor pathway inhibitor (TFPI) and the Z-protease inhibitor (ZPI). Activated factors VIII and IX greatly augment factor Xa production and this greatly enhances thrombin generation and fibrin formation. Thus, defects in factors VIII and IX are associated with impaired thrombin generation and clinically important bleeding.
|
|
|
What can be used to treat hemophilia?
|
Patients can receive cryoprecipitate to treat their hemophilia, which has factor VIII. There’s also a drug called DDAVP (Desmopressin) that is a natural hormone in the human body that regularly affects water regulation but causes levels of factor VIII to rise. DDAVP can be given intranasally or IV. Fresh frozen plasma can be used for factor IX.
|
|
|
What is the most common inherited bleeding disorder, and what is its inheritance pattern?
|
Von Willebrand Disease is the most common inherited bleeding disorder, and it has an autosomal recessive inheritance pattern.
Type 1 is the most common type and is characterized by a slight quantitative deficiency Type 2 is a qualitative deficiency (functional defect of protein) Type 3 is the most severe type and is characterized by a large quantitative deficiency |
|
|
Describe the different Type 2 vWDiseases.
|
Type 2A – either defective multimerization or more rapid proteolysis of the protein, with loss of HMW multimers (most important for platelet binding)
Type 2B – enhanced binding to platelet glycoprotein Ib, resulting in both HMW multimers and platelets being removed from circulation Type 2M – impaired binding to collagen or platelet glycoprotein 1b Type 2N (Normandy) – impaired binding of FVIII, resulting in FVIII deficiency |
|
|
What are the 2 important roles of vWF?
|
1) vWF binds platelets to the endothelium when injury occurs
2) vWF binds FVIII such that without vWF, you look like a severe Hemophilia A patient |
|
|
What cells synthesize vWF?
|
Megakaryocytes and endothelial cells synthesize vWF, and they are stored in Weibel-Palade bodies or alpha-granules in platelets.
|
|
|
How would you treat Von Willebrand Disease?
|
You could treat vWD with DDAVP (desmopressin) for mild disease which stimulates the release of vWF from endothelial cells and platelets.
You could also use a plasma-derived vWF concentrate for type 1 who don’t respond to DDAVP or need surgery or type 3 vWD. |
|
|
What is PAI-1, and what does it do?
|
PAI-1, synthesized by endothelial cells, is a serine protease inhibitor that inhibits tPA and uPA, which activate the conversion of plasminogen into plasmin for fibrinolysis. Therefore, PAI-1 is a plasminogen activator inhibitor.
|
|
|
What is PAI-2, and what does it do?
|
PAI-2 also inhibits tPA and uPA, but it is synthesized by the placenta, monocytes, eosinophils, and keratinocytes as well as by ovarian tumors and myeloid leukemic cells.
|
|
|
How many kringles does plasminogen have?
|
Plasminogen has 5 kringles.
|
|
|
What molecules inhibit plasmin?
|
• Serine protease inhibitors (AT< alpha2-antitrypsin)
• Alpha2-antiplasmin • Alpha2-macroglobulin • TAFI (thrombin-activatable fibrinolytic inhibitor) – also a plasminogen activator inhibitor |
|
|
What is DDimer?
|
DDimer is a fibrin degradation product that remains after the clot has been lysed. DDimer is used to tell whether or not thrombus formation and subsequent lysis had occurred. It’s used to diagnose:
• DIC • DVT • PE |
|
|
Why does bleeding occur in acute promyelocytic leukemia?
|
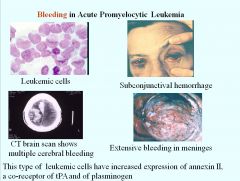
Leukemic cells in acute promyelocytic leukemia have increased expression of annexin II, which is a co-receptor for tPA and plasminogen. Plasmin gets formed, which lyses clots and induces bleeding.
|
|
|
How is uPA important for cell motility?
|
uPA activates plasmin, which activates metalloproteinases which breaks down the extracellular matrix. You have to break down the matrix so the cell can move. Plasmin also activates various growth factors and enhances cell proliferation.
In all tissue remodeling and wound healing, you need cell migration |
|
|
What are contraindications for thrombolytic therapy?
|

see slide
|
|
|
What causes of acquired coagulopathy are associated with both bleeding and clotting?
|
• Liver disease
• DIC • Drug-induced causes |
|
|
What are the procoagulants associated with Vitamin K? What about the anticoagulants?
|
Procoagulants associated with Vit K: Factors II, VII, IX, and X
Anticoagulants associated with Vit K: Protein C & S |
|
|
What are sources of Vitamin K?
|
• Intestinal flora (Abx + malnutrition can cause deficiencies)
• Diet: green leafy vegetables (severe malnutrition can cause deficiencies) |
|
|
What factor is first to be affected in liver disease, and why?
|
Factor VII would be the first affected in liver disease, because it has the shortest half life (4-6 hours).
|
|
|
What clotting factors does cryoprecipitate contain?
|
• Factors VIII, XIII,
• Fibrinogen • vWF • (It’s usually used to treat fibrinogen disorders) |
|
|
If you have a 70 year old woman enter your clinic with bleeding and bruising all over, and she says nothing like this has ever happened before, what would you suspect?
|
You may suspect Acquired FVIII inhibitor, the most common factor inhibited by a formed auto-Ab. You can perform a mixing study to see whether or not inhibitor is present and quantify the titer of Ab with the Bethesda assay.
|
|
|
What key steps of the coagulation cascade need phospholipids?
|
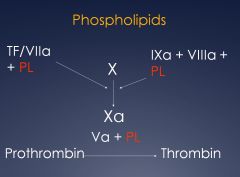
see image
|
|
|
In antiphospholipid syndrome, patients have clotting going in in vivo, but they have a prolonged PTT. Why is that?
|
Antibodies that form against the phospholipids include Lupus anticoagulant (LAC) and anticoardiolpin. LAC is a common cause of elevated PTT, although it doesn’t cause bleeding and is instead associated with thrombosis, although the mechanism is unclear.
|
|
|
If you have a 25 yo woman present with stroke, what acquired coagulopathy would be on your differential?
|
Antiphospholipid syndrome
|
|
|
How does antiphospholipid relate to pregnancies?
|
Antiphospholipid syndrome can cause consecutive loses of pregnancy either early or late in gestation. You would look for Lupus anticoagulant on the labs.
|
|
|
What is the most common cause of chronic DIC, and what is the source of complications?
|
Malignancy (cancer) is the most common cause of chronic DIC, and complications tend to be due to thrombosis.
|
|
|
Why would you see Vitamin K deficiency in liver disease?
|
You may see Vitamin K deficiency in liver disease due to decreased absorption of Vitamin K due to decreased bile salt production.
|
|
|
What are ways liver disease can predispose one to clotting?
|
In liver disease, you can have:
• decreased levels of AT III, protein C, and protein S • elevated heparin cofactor II • increased levels of vWF and FVIII |
|
|
What are ways liver disease can predispose one to bleeding?
|
In liver disease, you have:
• Decreased synthesis of coagulation factors (2, 5, 7, 9, 10, 11) • Decreased absorption of Vitamin K • Decreased fibrinogen synthesis and qualitative problems with fibrinogen • Hypersplenism can cause platelet sequestration (thrombocytopenia) • Platelet inhibition by NO and prostacyclin |
|
|
What is the risk for thrombosis with Antiphospholipid syndrome, and how would you diagnose the illness?
|
The risk for thrombosis is increased with antiphospholipid syndrome, and diagnosis could be:
• Clinical o Vascular thrombosis (one or more in artery, vein, or small vessel in any tissue or organ) o Pregnancy 1 or more death of morphologically normal fetus after 10th gestation OR 3 or more premature births of morphologically normal neonates at or before 34th gestational week OR 3 or more unexplained consecutive spontaneous abortions prior to the 10th gestational week • Laboratory o Anticardiolipin Ab: IgG or IgM o Lupus anticoagulant Ab on 2 or more occasions >=6 weeks apart |
|
|
What is Virchow’s triad?
|

Stasis
Vessel wall injury Hypercoagulability |
|
|
What risk factors can increase the propensity for a DVT and a PE?
|
The following can increase one’s risk for thrombosis:
• Overweight/Obese • Use of Transdermal Estrogen (used for menopause) • Oral contraceptives |
|
|
What are some differences between arterial and venous thrombosis?
|
Arterial thrombosis is platelet driven and often associated with atherosclerosis.
|
|
|
What is adiponectin, and what does it do?
|

Adiponectin is a hormone that has effects on liver and muscle and is an inhibitor of platelet aggregation. When someone gets obese, their adipocytes get engorged with fat, and the chemical composition of the adipocyte changes. The hormone leptin is released, and it tells the brain not to eat so much. Leptin increases the tendency of platelets to aggregate (has pro-thrombotic activities).
|
|
|
What’s a common cause for the formation of thrombi in the left iliac vein?
|
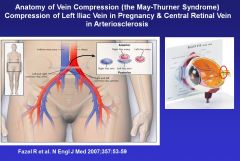
During pregnancy, the expanded uterus can cause the right iliac artery to compress the left iliac vein and cause venous stasis. Whenever you see a pregnant woman with a swollen left leg; it’s indicative of this process.
|
|
|
Besides left iliac compression, what’s an anatomical example of thrombus formation?
|
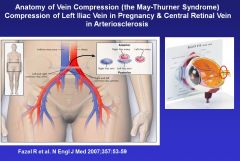
There’s a cribriform plate in the back of the retina that the retinal artery and retinal vein pass through. In people with HTN, the retinal artery can get increased flow and an increased diameter and consequently compress the retinal vein, and the blood forms a thrombus due to stasis->can cause loss of vision.
|
|
|
Mutation in what molecule presents the highest relative risk for thrombosis?
|
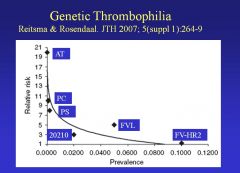
• Anti-Thrombin (AT) – uncommon mutation
Followed by: • PC • PS • FVL – common, 5% risk • 20210 (area of pro-thrombin molecule) – common but low risk |
|
|
How does Factor V Leiden cause thrombosis?
|
• Protein C cleaves activated FV at Arg residues. Factor V Leiden has a mutation where one of the arginines is changed to glutamic acid (base 1691 at position 506), rendering FVa relatively resistant to cleavage. Factor Va stays around a little longer than normal, which increases the risk for thrombosis.
• Protein C also cleaves Factor VIII, but it needs a cofactor, and normal FV is a cofactor for protein C, so it doesn’t cleave FVIIIa either. |
|
|
Why is FVL as prevalent as it is?
|
Incidence of intracranial hemorrhage in newborns is lower in those with FVL. This could help explain the preponderance of FVL mutations given its deleterious effects.
|
|
|
What happens in the Prothrombin 20210 mutation?
|
The 20210 mutation causes an increased production of prothrombin so that the thrombosis risk parallels the prothrombin concentration. They have 30-50% more prothrombin, which stimulates more thrombin generation in the individuals. Thrombin activates TAFI.
TAFI inhibits fibrinolysis, which increases risk for thrombosis |
|
|
What is the Prothrombin G20210A mutation, and in what population does it most frequently occur?
|
Prothrombin G20210A occurs most frequently in the Spanish population and increases the risk of 2nd/3rd trimester fetal death. It manifests as DVT/PE, cerebral venous thrombosis, and peripheral artery disease.
|
|
|
Why must you screen newborns for Protein C and Protein S deficiency?
|
If a baby is homozygous for Protein C and Protein S deficiency, they begin to thrombose hours after birth, and purpura fulminans occurs, which is lethal if not treated with Protein C and Protein S.
|
|
|
What is the relationship between Protein S and estrogens?
|
Protein S circulates 1/3 free and 2/3 bound to 4th component of complement binding protein. Free Protein S is decreased by estrogens (by causing increased binding to C4BP), pregnancy, and is low in SCD and HIV infection.
|
|
|
What deficiencies can lead to hyperhomocystenemia, and what is the clinical significance?
|

• Deficiencies in MTHFReductase, B12 & Folate, Cystathione Synthase-B6.
• The clinical significance for increased homocysteine is a greater propensity for strokes, heart attacks, and venous thrombosis. |
|
|
When do you investigate for thrombophilia?
|

FURY
|
|
|
What is the mechanism of action of warfarin?
|
Warfarin inhibits the reduction of Vitamin K so that it cannot serve as a cofactor for the clotting factors. (prevents carboxylation). The clotting factors can’t be carboxylated at their glutamic acid residues and are incapable of binding to the endothelial surface of blood vessels and are thus rendered inactive. Warfarin also inhibits protein C and protein S due to inhibition of carboxylation.
|
|
|
Which proteins are carboxylated?
|
Proteins carboxylated are II,VII,IX,X,C,S,Z, osteocalcin, matrix Gla,Gas6
|
|
|
Why may you not use warfarin in an emergent situation?
|
Warfarin is slow-acting and requires 5 days of dosing to achieve therapeutic INR; thus you probably would not use it for an emergency thrombotic event.
|
|
|
What is the desired INR for Warfarin therapy?
|
INR 2-3
|
|
|
What is warfarin necrosis, and how does it occur?
|
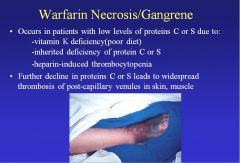
Warfarin necrosis occurs in those who have low levels of protein C or S, because warfarin decreases the levels of protein C and S, which are responsible for thrombolysis. The result is severe thrombosis.
|
|
|
What are the drug interactions for warfarin?
|

see slide
|
|
|
What is the mechanism of action of heparin?
|
Heparin increases the activity of Anti-Thrombin (AT) 1000 fold, and AT binds thrombin and renders in inactive. It has a 5 unit saccharide-site that binds the AT. Longer chains also bind thrombin which brings the thrombin directly into contact with the AT, which potentiates the effect. Heparin can also inhibit FactorXa (which directly binds AT and gets inactivated by AT).
|
|
|
What are advantages and disadvantages of heparin?
|
Advantages:
1. It works immediately 2. It’s very safe and effective Disadvantages: 1. You can’t give heparin orally, because it gets inactivated by the gut. 2. Heparin-AT complex inhibits F9a, 10a, 12a, 7a-TF and thrombin 3. Inhibits smooth muscle cell proliferation and angiogenesis 4. It can bind a variety of plasma proteins and become inactivated 5. 90 min half-life (short) 6. HIT (Heparin-Induced Thrombocytopenia) |
|
|
How would you administer heparin to prevent thrombosis? What would you use to stop heparin?
|
• 5-10,000 U sc every 8-12h
• To stop heparin, administer protamine (readily reversed). |
|
|
What is the use and purpose of Low Molecular Weight Heparins? What’s a disadvantage?
|
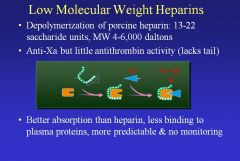
• LMWH have anti-Xa but little AT activity due to the lack of the tail. It has better absorption (smaller) and less binding to plasma proteins, making it more predictable and reducing the need for monitoring. And it’s just as effective as heparin.
• A disadvantage is that protamine doesn’t work as well for reversing LMWH, so bleeding is a risk. |
|
|
What is Fondaparinux (Arixtra) and what is it used for?
|
Fondaparinux has a synthetic 5 saccharide binding site of AT and has only Xa activity. It’s at least as effective as LMWH for prophylaxis.
• It has 90% bioavailability after s.c. injection. • Mininal protein binding, no monitoring needed • T1/2 17 hours; once daily injection • Minimal interaction with platelets (no HIT) • Equivalent to IV UH in efficacy with less bleeding for PE • Equivalent to 2x daily LMWH for DVT Adverse: • It’s entirely excreted by the kidney, so you can’t give it to patients with kidney failure. • No reversal agent |
|
|
What are advantages of Rivaroxaban (Xarelto)?
|
• It’s a direct Xa inhibitor and can be given by mouth. It does not require AT.
• It’s approved to treat AF. |
|
|
What is hirudin, and how does it work? What are its derivatives?
|
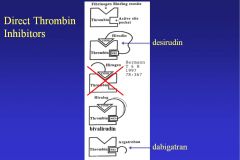
Hirudin is a molecule used by leeches to bind and prevent thrombin from converting fibrinogen to fibrin. It’s approved to treat HIT and given by IV infusion. Also, it’s monitored with aPTT (range: 1.5-2.5 times control).
It works by blocking the binding site for fibrinogen on thrombin. Bivalirudin binds both the fibrinogen site and the active site of thrombin. Argatroban and dabigatran just bind to the active site. |
|
|
What are adverse effects of Lepirudin?
|
Adverse effects:
Bleeding is main adverse effect. Antihirudin Abs in 40% of treated patients, usually seen after 8-10 days of dosing. Renal clearance impaired, enhancing anticoagulant effect. Switching to warfarin is problematic, because lepirudin prolongs the PT. |
|
|
What is the drug profile for argatroban?
|
• Synthetic direct thrombin inhibitor
• Reversibly binds to catalytic site of thrombin • Given IV for HIT; steady state in 1-3 hrs • Half-life is 39-51 min; return to baseline in 2-4 hrs • Monitor with aPTT • Conversion to warfarin problematic because argatroban prolongs prothrombin time |
|
|
What is the drug profile for Dabigatran?
|
• Oral prodrug, converted to dabigatran, a small molecule direct & reversible thrombin inhibitor
• Half-life 14-17 hr, eliminated by the kidneys • Dose: 75 or 150 mg bid, depending on bleeding risk • Food has minimal effect on absorption • Does not require monitoring; more convenient & at least as effective as warfarin • Approved to prevent thrombosis in patients with atrial fibrillation |
|
|
What is apheresis?
|
Apheresis is the process by which donor blood flows into a centrifuge to skim off platelets and return the RBCs and plasma. Plasma and granulocytes can also be collected by apheresis.
|
|
|
What is an allogeneic blood donation?
|
An allogeneic blood donation is from someone else (non-self), as opposed to an autologous donation, which would be a patient donating blood before a surgery for transfusion back to themselves afterwards.
|
|
|
In an acute hemorrhage, how much volume loss would be needed before the patient would require RBCs?
|
30-40%. Volume replacement would be done first by crystalloids.
|
|
|
If uncrossmatched blood were to be given, from which ABO blood group would it be received (A, B, AB, O)?
|
Uncrossmatched group O RBCs can be given to almost any patient.
|
|
|
If a patient can compensate, what’s the Hb cutoff for what can be tolerated, and at what point does mortality start?
|
Hb >7-8 g/dL can be tolerated, and mortality starts at Hb<=5g/dL
|
|
|
What is the goal for post-transfusion Hb?
|
The goal is <= 10 g/dL Hb
|
|
|
In normovolemic anemia, how do patients compensate such that they maintain relatively normal O2 delivery and consumption?
|
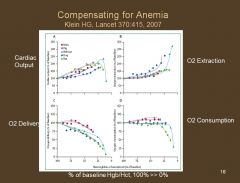
They increase Cardiac Output (CO) and O2 extraction.
|
|
|
What’s the shelf life of blood transfusion units, and how much of a rise in Hb does one unit correspond to?
|
Blood transfusion units have shelf lives of 42 days, and for the average adult, 1 unit corresponds to 1 g/dL rise in Hb.
|
|
|
How long can platelets be stored for?
|
Platelets can be stored for up to 5 days.
|
|
|
At what point would you transfuse platelets to a patient (how low would there platelets have to be)?
|
Transfusion of platelets is at a cutoff of 10,000 platelets in the patient.
40-50,000 for a surgery or invasive procedure. |
|
|
What rise in platelets would you expect after a platelet transfusion of 1 unit?
|
You would expect a 30,000/uL rise in platelets after a 1 unit transfusion just afterwards with a circulating half life of 3-4 days.
|
|
|
What could be reasons for low response to platelet transfusion?
|
• Larger patient, higher blood volume
• Increased platelet turnover o Bleeding, infection, fever, DIC • Platelet sequestration (splenomegaly) • Ab-mediated destruction due to autoantibodies (ITP) or alloantibodies (HLA) |
|
|
When would plasma be transfused into a patient?
|
• Plasma would be transfused to replace soluble clotting factors. It’s not given prophylactically, as might be done for platelet. It may be given for bleeding or during an invasive procedure.
• Warfarin reversal or Vitamin K deficiency (in emergent conditions) • TTP |
|
|
How does plasma transfusion help TTP patients?
|
TTP patients have large hyperactive vWFs and low levels of ADAMTS13, which cleaves vWF. Plasma exchange provides the ADAMTS13 so that the vWFs can be cleaved, and the patient would have normal platelet hemostasis.
|
|
|
Which clotting factors have the shortest circulating half-lives?
|
F7 (8 hours) and F8 (12 hours) have the shortest circulating half-lives.
|
|
|
What type of plasma could you give to almost anyone?
|
You could give AB plasma to almost anyone. Otherwise, it should be ABO-compatible.
|
|
|
What is cryoprecipitate used for?
|
• Cryoprecipitate is used as a source of fibrinogen for people with low fibrinogen (liver disease, DIC)
• Platelet dysfunction (vWF) |
|
|
When would granulocytes be transfused?
|
Granulocytes can be collected by apheresis and transfused to treat serious refractory bacterial and fungal infections, as an adjunct to Abx in severely neutropenic patients. Benefit difficult to evaluate due to low WBC content of traditional products being relatively low.
|
|
|
Are the Abs made for ABO Ab reactions IgG, IgM, IgA, or what? What about for Rh Ab reactions?
|
• Abs made in ABO immune responses are IgM
• Abs made in Rh immune responses are IgG |
|
|
What is Acute Hemolytic Transfusion Reaction (AHTR), and what are its characteristics?
|
• AHTR occurs in 1 in 75k RBC units. Intravascularly, IgM binds RBC, fixes complement and lyses RBCs
• RBC membranes go and damage the kidney – acute tubular necrosis • Immune complexes and thrombotic ischemia occurs w/ cytokine activation |
|
|
What Ig mediate the allergic reactions in blood transfusions? What about the anaphylactic reactions?
|
Allergic – IgE
Anaphylactic – IgA |
|
|
What is Transfusion-Related Acute Lung Injury (TRALI), what is the main mechanism, and what are some of its characteristics?
|
• TRALI occurs in about 1 in 5000 units when the donor’s Abs attack the patient’s WBCs
• Patient presents with SOB, hypoxia, pulmonary edema • There could be fluid overload, a cardiac event • Has 5-10% mortality and no specific therapy for it • Most common occurs from pregnant/women donors (they have more HLA and WBC Abs), so most plasma comes from men |
|
|
How do you prevent WBC transfusion Graft Versus Host Disease (GVHD)?
|
You can gamma-irradiate the RBCs, platelets, granulocytes, which kills the WBCs/lymphocytes, but doesn’t bother the RBCs.
|
|
|
What causes febrile transfusion reactions?
|
WBCs in the blood product can cause febrile transfusion reactions either by being targets for WBC Abs in the patient or by generating pyrogenic cytokines like IL-1 and TNF during blood storage. The differential may include acute hemolytic reaction and septic reaction (bacterial contamination in blood bag). Antipyretics and meperidine are useful therapies.
|
|
|
What are Delayed hemolytic transfusion reactions (DHTR), and which type of test would you use for DHTR, direct coombs or indirect?
|
DHTRs occur when transfused RBCs stimulate an anamnestic Ab response in a previously immunized patient. The incompatible RBCs are coated with Ab and gradually destroyed via extravascular hemolysis. Direct test for Abs bound to RBCs.
|
|
|
When is the reticulocyte count increased? Decreased?
|
Reticulocyte count is increased in…
* Accelerated RBC production (e.g., recovery from blood loss) * Accelerated RBC destruction (e.g., hemolytic anemia) Reticulocyte count is decreased in… * iron Deficiencies * Vitamin B12 & folic acid deficiencies * Anemia of chronic inflammation (AI) |
|
|
When would you see a decrease in haptoglobin?
|
Haptoglobin complexes with Hb and is phagocytosed and degraded by macrophages. When there is an excess of Hb in the serum (e.g., intravascular hemolytic anemia), then there would be a decrease in haptoglobin.
|