Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
267 Cards in this Set
- Front
- Back
|
List the 5 roles of liver function
|
1) storage of iron, carbohydrates, vitamins
2) filtration & storage of blood 3) metabolism of carbohydrates, proteins, fats, chemicals & toxins 4) formation of bile 5) formation of coagulation factors |
|
|
List the features of a lobule
|
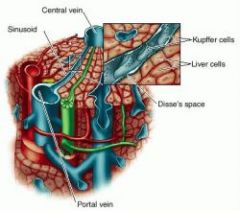
1) portal vein -> sinusoids bathing Kuppfer cells lining Space of Disse surrounding hepatocytes and draining into central vein
2) Space of disse drains into lymphatic duct 3) Bile canniculi, draining from hepatocytes -> bile duct |
|
|
what is the feature of vascular resistance through the liver and how is it clinically relevant
|
very low pressure gradient. Cirrhosis or blockage of portal vein -> capillary leakage into intestines (portal hypertension) -> rapid fluid loss, possible death.
|
|
|
what quantity of blood does the liver store and what is the conswequence of RVF
|
Holds 0.5L but with RVF can hold up to a litre: smooth & distended.
|
|
|
what is the role of the liver in carbohydrate metabolism
|
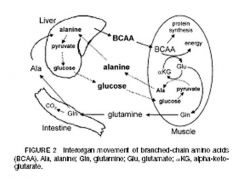
1) amino acids (via alanine shuttle), lactate and glycerol -> gluconeogenesis
2) glycolysis 3) pentose phosphate pathway 4) lipogenesis |
|
|
Action of secretin?
|
HCo3- production by pancreatic duct and duodenum Brunner's Cells
|
|
|
What are the effects of CCK
|
Intestinal phase of digestion:
1) Inhibits gastric emptying & H+ secretion 2) Potent contracter of gall bladder and relaxation of Sphincter of Oddi 3) Duodenal digestion: release of pancreatic lipase, digestive enzymes and bicarb. |
|
|
GIP
|
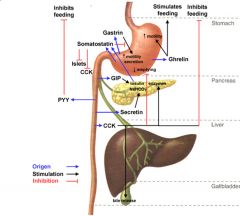
Acts when carbohydrate increases in gut to increase insulin secretion
|
|
|
What are the effects of VIP
|
1) Inhibition of H+ production
2) Increased bicarbonate & electrolyte secretion secretion |
|
|
Describe the differences between the preganglionic & postganglionic fibres of SNS and PNS
|
PNS long pre fibre, short post fibre, ACh released at ganglion with SNS & PNS, SNS post ganglionic fibres long. Post ganglionic fibres are slow (unmyelinated)
|
|
|
What sense receptors are present if a stimuilus occurs in the gut
|
mechano, chemo receptors of local afferents (enteric), and SNS, endocrine (increased or decreased motility, increased secretions (activated by gastrin, VIP))
|
|
|
What are the 2 main functions of an enteric plexus neuron
|
sensory (osmotic, chemical, stretch)
Motor and interneurons |
|
|
What is the route of most sympathetic nerve fibres
|
along blood vessels because of vasoconstriction of blood vessels
|
|
|
What are the effects of CCK
|
Following lipid meal: decreases gastric emptying, increases lipase secretion, increases gall bladder contractions, increases insulin secretion
|
|
|
Describe a typical proton pump
|
H+/K+ ATPase
|
|
|
What receptors are present if some stimuilus occurs in the gut
|
mechano, chemo receptors of local afferents (enteric), and SNS
|
|
|
What types of neurons are there in the submucosal and myenteric plexus
|
Both involved in:
sensory (osmotic, chemical, stretch) Motor (contraction, glandular secretions) and interneurons |
|
|
What is the route of most sympathetic nerve fibres
|
along blood vessels because of vasoconstriction of blood vessels
|
|
|
CCK
|
Lipid meal: decreases gastric emptying, increases lipase secretion
|
|
|
What is the proton pump
|
H+/K+ ATPase
|
|
|
List the stimulators of H+ release into the stomach
|
Gastrin, AcH, histamine, decrease in prostaglandins and somatostatin
|
|
|
What is the pathway of somatostatin and prostaglandin inhibition of H+
|
stimulates G inhibitory protein, inhibits adenylate cyclase, decreased cAMP, decreased phosphorylation of protein kinase, decreased activity of ATP dependent H+/K+ pump
|
|
|
What are the 3 classes of enteric plexus neurons
|
1) sensory
2) motor 3) interneurons |
|
|
What are the motor functions of the enteric nerve plexuses
|
1) motility (2) secretions (H+, pepsinogen, mucous, endocrine cells
|
|
|
What sensory functions are there of the enteric plexuses
|
1) mechano (2) osmotic (3) chemo (glucose, amino acids)
|
|
|
Describe the features of the PNS innervation of the GI tract
|
1) vagal (brain stem) and pelvic (S2-4) with no branching - very localised
2) ganglionic neurons in ENS 3) excitatory: contractions, secretions, hormones, vasodilation |
|
|
Describe the features of the SNS innervation of the GI tract
|
1) postganglionic fibres from foregut (coeliac), midgut (superior mesentary), hindgut (inferior mesentary - very widespread effects
2) wide inhibitory effects |
|
|
What is meant by humoral motility
|
1) gastrin: increased motility
2) motilin: migrating motor complex 3) CCK: decreased gastric emptying when fat detected in chyme. |
|
|
What are the effects of 4 hormones that regulate H+ secretion in the stomach
|
1) Distension -> gastrin (G-cells in antrum) -> G+ protein -> 2nd messenger -> +H+/K+ pump
2) glucose or fat in duodenum -> GIP (K-cells) or CCK (I-cells) -> inhibition of H+ 3) high pH ECL cells (antrum) -> Histamine: stimulates G+ protein -> cAMP -> +H+/K+ pump 2) low pH -> somatostatin (D-cells in antrum), prostaglandins activate G- inhibitory protein -H+/K+ pump |
|
|
Explain the consequences of G6Pase deficiency
|
1) pushes glycolysis -> pyruvate -> acetyl CoA -> lipogenesis (hypertriglyceridemia, lactic acidosis, fatty liver)
2) production of oxaloacetate inpaired by inhibition from high G6P -> no gluconeogenesis even at high levels of glucagon 2) pentose phosphate pathway -> purines -> uric acid accumulation 3) G6P -> G1P -> glycogenesis -> fatty liver & kidney 4) chronic hypoglycemia due to entrapment of G6P, increased glycogenesis and lipogenesis -> prolonged elevated cortisol -> muscle wasting, osteomalacia etc. |
|
|
How is short term hypoglycemia different to long term
|
1) short term: SNS (glycogenolysis stimulated by adrenalin in muscle) and glucagon in liver, glucagon inhibits insulin release).
2) long term: glucagon, cortisol, growth hormone -> gluconeogenesis from AAs, lactose and glycerol, production of ketone bodies from lipids, B-oxidation of fatty acids |
|
|
What triggers cause insulin release
|
1) high blood glucose
2) glucose in gut stimulates GIP -> increased insulin secretion (incretin effect) 3) AAs in gut stimulate gastrin and CCK -> stimulate insulin (incretin effect) |
|
|
List the compensatory mechanisms for pH balance arising from diarrhoea
|
1) Diarrhoea -> loss of HCO3- -> metabolic acidosis
2) Equation CO2 + H2O <> H+ + HCO3- 3) Rapid response: Equilibrium shifts to left, reducing decrease in blood pH 4) medium term response: rapid respiration to shift equilibrium further to the left 5) slower response: increased renal excretion H+ in the proximal tubules & collecting ducts (alpha intercalated cells) |
|
|
What is effect on pCO2 caused by chronic diarrhoea
|
1) Diarrhoea -> loss of HCO3- -> metabolic acidosis
2) Equation CO2 + H2O <> H+ + HCO3- 3) Equilibrium shift to right decreases pCO2 |
|
|
List the starting & end products (enzymes?) & energy requirements of gluconeogenesis
|
1) glucose -> G6P (hexokinase)
2) g6P -> glycogen (1ATP, 1UTP) |
|
|
What hormones stimulate glycogenolysis
|
1) adrenaline in muscle
2) glycagon in liver |
|
|
which hormones affect gluconeogenesis
|
1) insulin inhibits
2) glucagon and adrenaline activate |
|
|
Which prostaglandins are involved in inhibiting cAMP production in parietal cells
|
PGE2
|
|
|
What are the 3 phases of gastric secretion
|
Cephalic (parasympathetic vagus -> increased gastric secretions - chief, parietal, G, ECL cells)
Gastric - distension of stomach, chemical (proteins), neural and hormonal (histamine, gastrin) Intestinal phase (duodenum receives chyme with fat: CCK, secretin, GIP, decrease stomach motility and secretions, release of bile, pancreatic enzymes and bicarb, increased intestinal motility |
|
|
What cells produce what hormones in the stomach & duodenum
|
1) somatostatin - D cells, inhibits everything, especially H+ (pyloric antrum)
2) gastrin - G cells below gastric glands, stimulated by stretch increases motility and H+ 3) Secretin: S-cells within duodenum, bicarb (pancreatic duct cells and Brunner's in submucosa 4) CCK: I-cells in duodenum: bile secretion and pancreatic enzyme production, appetite inhibition 5) GIP: K-cells in duodenum, glucose sensitive, secretion of insulin, inhibits gastric secretions 6) VIP: from nerve endings, stimulates motility and electrolyte secretions |
|
|
What are the 3 types of antagonism
|
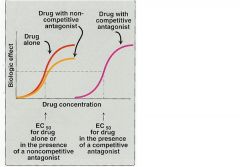
compeditive (competes witth substrate for active site)
non-compeditive (binds to allosteric site) un-compeditive (binds only when substrate is bound) |
|
|
Where is most of glycogen stored and how is it utilised
|
1) skeletal muscle
2) muscke doesn't have G6Pase, so glucose cannot be released to the bloodstream |
|
|
Describe GLUT1
|
ubiquitous
|
|
|
Describe GLUT 2
|
1) basolateral intestine
2) pancreas 3) liver |
|
|
Describe GLUT3
|
everywhere
|
|
|
Describe GLUT4
|
muscle, adipose tissue
|
|
|
Describe GLUT5
|
brain
|
|
|
List the effects of insulin
|
glucose uptake in skeletal muscle, FA uptake in adipose, increased glycolysis, glycogenesis, lipogenesis, inhibits - B-oxidation, glycogenolysis, gluconeogenesis
|
|
|
Describe B oxidation
|
1) Acyl-CoA carnitine transported across inner membrane of mitochondria
2) Oxidation 1 produces FADH2 from FAD (produces double bond) 3) Hydration of double bond 4) Oxidation to produce NADH 5) cleavage between both =O plus another CoA produces acetyl CoA (can accumulate) plus shorter FA |
|
|
What are the 2 types of amino acids
|
glucogenic
ketogenic (lysine, leucine) |
|
|
List the organs that solely use glucose as fuel
|
Brain, testes, erythrocytes, renal medulla
|
|
|
Describe the energy requirements of gluconeogenesis (describe the ATP/GTP/NADH utilising steps)
|
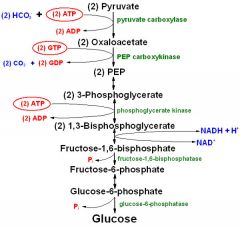
2 ATP (pyruvate carboxylase (in mito adds 2xCo2 to 2xpyruvate - 2xOAA)
PEP carboxykinase (removes 2xCO2 from 2 x OAA using 2 gtp - irreversible) 2x3 phosphoglycerate -> 2x1,3 DPG (recall inhibition by 2-3 DPG) + 2xATP 2x1,3bisphophoglycerate -> fructose-1,6-bisphosphate (1 NADH) Total = 4ATP, GTP, NADH to make 1 glucose molecule |
|
|
List the major fuel inputs of gluconeogenesis
|
glycerol, lactate, pyruvate, oxaloacetate, amino acids (primarily alanine & glutamine)
|
|
|
List the 3 key substrates used to transfer 2 carbon units between the cytosol and mitochondrial matrix
|
cytosol: pyruvate > oxaloacetate > citrate (Acetyl CoA > CoA)
Reverse is true for mitochondrial matrix |
|
|
What does the formation of malonly CoA signify. How is its formation regulated?
|
1) Committment to fatty acid synthesis of acetyl CoA because Acetyl CoA Carboxylase reaction is irreversible
2) Insulin & citrate signals abundance. Insulin causes dephosphorylation of ACC, activating it. AMP stimulates protein kinase to phosphorylate ACC (inactivation), signalling ATP depletion. |
|
|
What is required for De Novo synthesis of fatty acids
|
Acetyl CoA for first step reaction to malonyl CoA, then NADPH
|
|
|
Where in the cell do fatty acid synthesis and B-oxidation occur
|
Synthesis: cytosol
B-oxidation: mitochondria |
|
|
List 2 sources of glycerol
|
1) glycolysis (DHAP)
2) TAG breakdown |
|
|
Describe the fate of glucose absorbed by adipocytes
|
G > G6P > pyruvate > AcCoA -> fatty acid synthesis (some goes to TCA)
|
|
|
Describe the fates of glucose in the liver
|
1) G > G6P then
1) glycogenesis 2) glycolysis > pyruvate (or lactate if anaerobic) 3) pyruvate -> AcCoA -> TCA 4) AcCoA ->fatty acid synthesis 3) Pentose phosphate pathway > NADPH (for fatty acid synthesis) + pentose for nucleotides |
|
|
What is metabolic syndrome
|
Cluster of hypertension, dyslipidemia & insulin resistance
|
|
|
List the co-morbidities of obesity in men
|
1) decreased testosterone due to increased aromatase
2) Metabolic Syndrome: hypertension + hyperlipidemia > cardiovascular disease 3) insulin resistance > T2 diabetes > more co-morbidities 4) respiratory - sleep apnoea, |
|
|
Describe the cephalic stage of digestion
|
1) Cerebral cortex + hypothalamus stimulates medulla > vagus nerve > post-ganglionic neurons ->
1) direct stimulation of parietal cells + ECL cells -> release histamine -> H+ secretion 2) release of gastrin from G cells -> enhance H+ secretion 3) synapses with myenteric plexus (between middle circ and outer long muscle layers) increasing stomach motility |
|
|
Describe the gastric phase of digestion
|
1) Stretch receptors increase contractility & gastric emptying
2) Stimulagion of G-cells to produce gastrin, ECL cells to secrete histamine > acid, pepsinogen, intrinsic factor release |
|
|
Describe the mechanisms that inhibit appetite
|
1) Gastric stretch > vagal feedback
2) Fat in duodenum > CCK > rapid hypothalamus downregulation of appetite 3) Inhibition of neuropeptide Y by leptin 4) Decreased ghrelin (P/D1 cells of fundus) 5) Increased glucose & fatty acids > increased insulin |
|
|
Describe the intestinal phase of digestion
|
1) Acidic chyme enters duodenum > secretin > pancreas & Brunner cells HCO3
2) Fat entering duodenum > CCK > pancreatic lipases + gall bladder contraction 3) Feedback: secretin + CCK inhibit gastric emptying, contractility, secretions |
|
|
How does binge eating disorder differ from bulemia
|
No compensatory mechanism (bulemia can compensate by purging: vomit or diarrhoea)
|
|
|
What are the cognitive-behavioural features of ages 10-13
|
1) concrete thinking
2) peer group adherence |
|
|
What are the features of teen ages14-16
|
1) volatile mood swings
2) narcissistic, egocentric |
|
|
How is adolescent maturity measured on a scale of I-V
|
Tanner scale I-V based on
1) development of testes and breasts 2) pubic hair characteristics |
|
|
Describe the deamination of glutamate, why is it important
|
Glutamate > alphaketoglutarate (+NADH) + NH3
Only way to remove nitrogen |
|
|
What are the components of irreversible hypovolemic shock
|
hypovolemic: venous return falls because of blood loss > decreased CO > decreased perfusion (affects CNS > neurogenic shock enters, affects myocardium > cardiogenic shock enters) > anaerobic respiration, insufficient ATP to drive ion pumps, lactic acid release > membrane failure, cell lysis K+, Ca+ release > cascading tissue necrosis > massive inflammation > capillary leakage(distributive shock enters) > hypovolemia > organ & heart failure > death
|
|
|
What is the mechanism behind fluid replacement in shock (what types of shock?)
|
Fluid replacement increases venous return > increased CO (increased SV by Frank-Starling) > increased MAP > increased tissue perfusion (nutrients & oxygen)
|
|
|
Describe the mechanism of compensated shock
|
1) Frank-Starling: hypovolemia (hypovolemic or distributive shock) > decreased EDP > lower myocardial stretch > decreased SV compensated by increased HR > CO maintained by CO = SV x HR
2) baroreceptors (aortic arch, carotid sinus) sympathetic activity increases HR & contractility further increasing CO & ABP 3) increased PR by sympathetic + local vasoconstriction around site of blood loss 4) Renal effects: vasopressin & angiotensin II vasoconstriction increases MAP (from ohms law MAP = CO x TPR) |
|
|
What are the short term shock compensatory mechanisms
|
1) Frank-Starling
2) SNS (HR, contractility, arteriole constriction) |
|
|
A person comes to ED an hour after an MVA with shock. What are the medium term compensatory factors
|
1) Renal: renin secretion (AGII > aldosterone (fluid retention)
2) Hypothalamus osmoreceptors: vasopressin (vasoconstriction, decreased renal output) |
|
|
In shock management, why must fluid be added carefully
|
1) Rapid shift between fluid compartments (intravascular > interstitial > intracellular > fluid overload & oedema
2) reperfusion injury: mobilisation of clots, reactive oxides ( peroxides, superoxides), potassium, calcium & H+ leading to widespread membrane destabilisation and cell damage |
|
|
How does ethanol contribute to and complicate treatment of MVA casualties
|
1) impaired sensory, judgement performance, increased euphoria and confidence > risky behavious
2) shock management: increases arteriole vasodilation, decreases clotting, increases diuresis (inhibits ADH secretion) > negates compensatory mechanisms 3) aggression, decreased level of consciousness inhibits effective treatment |
|
|
What is pulmonary wedge pressure and how can it differentiate between types of shock
|
PWP: pulmonary artery is occluded and the distal pressure is same as left atrial pressure
1) if PWP (left atrial pressure) is low > decreased venous return > distributive or hypovolemic shock 2) if PWP is high > venous return is too high > left ventricular failure or mitral regurgitation > cardiogenic or neurogenic shock |
|
|
What are the possible consequences of haemopneumothorax
|
1) blood volume loss > hypovolaemic shock
2) pneumothorac > filling pleural cavity > collapse of lung > decreased alveolar area > hypoxia > exacerbation of tissue hypoxia and severity of shock |
|
|
What are the clinical signs of haemopneumothorax and explain the mechanisms
|
1) hypoxia > lip, tonguen nail cyanosis compensated partly by increased RR to increase O2 and decrease CO2
2) hypovolemic shock compensatory mechanisms: vasconstriction (pallor, cold), sweating (SNS vasoconstriction) increase HR (SNS, Frank-Starling) but increase SNS sweating, decreased or nil urine output |
|
|
Virchow's triad
|
Pulmonary embolism (triad = blood stasis, endothelial damage, hypercoagulation)
|
|
|
How does glucagon stimulate lipolysis
|
1) increase hormone sensitive lipase in adipose -> FFA
2) increase lipoprotein lipase in VLDLs to deliver newly synthesized FFAs |
|
|
List the possible target areas of insulin resistance
|
1) insulin receptor expression down regulation - decreased tyrosine kinase activity
2) impairment of GLUT 4 translocation 3) impaired signalling pathways |
|
|
What is a key indicator of insulin resistance
|
BMI indicating obesity (30+)
|
|
|
How is appetite regulated and how may obesity occur
|
1) Leptin decreases appetite acting on receptors in hypothalamus, increases insulin sensitivity
2) Neuropeptide Y stimulates appetite (ovexpressed in obese individuals - may be related to increased glucocorticoids) 2) Positive feedback with overeating - more fat, decreased leptin, greater appetite -> more fat 4) High levels of circulating FFA -> insulin resistance -> hyperglycemia -> diabetes |
|
|
What are the exocrine secretions of the pancreas (and what cells secrete them)
|
1) ductal centroacinar cells: bicarbonate &amp; water
2) acinar cells: digestive enzymes (trypsin and chymotrypsin, lipase, amylase, nuclease) |
|
|
List the 4 states of decreased consciousness
|
1) drowsiness (easily arouse)
2) stupor (vigorous arousal) 3) coma (non-arousal, bilateral withdrawal to pain, eyes closed) 4) vegetative (bilateral extension to pain, eyes open) |
|
|
What are the 3 criteria for Glasgow Coma Scale
|

1) Eyes (open, respond to sound or pain, closed)
2) Voice (conversation, confused, no noise) 3) motor (command, withdrawal, flexion, extension, nil) |
|
|
List the 5 types of diarrhoea
|
Exudative (blood, mucus)
secretory (serous) osmotic malabsorptive deranged motility (hypermotility - decreased transit time) |
|
|
What is the primary concern of uncontrolled secretory diarrhoea
|
vascular fluid loss -> hypovolaemic shock
|
|
|
What types of diarrhoea can be treated with antidiarrhoeals
|
Malabsorptive and deranged motility. (secretory, exudative, osmolar will be unaffected by decreasing peristalsis)
|
|
|
How can a "fat: child still be malnourished
|
High fat and CH intake replaces
1) essential amino acids and fatty acids 2) enzyme cofactors (vitamins, minerals) |
|
|
List the steps of ox-phos
|
1) Electrons passed from NADH via proton pumps (complexes 1,3,4) and intermediate electron carriers (CoQ, CytC)
2) Protons pumped into space between inner & outer membranes 3) ATP synthase uses H+ gradient across inner membrane to convert ADP to ATP in matrix. |
|
|
What are the 2 ways B-cells and the liver are prevented ffrom "stripping" the blood of all glucose
|
1) Glucokinase hs high Km, hence high conc. of extracellular glucose required to trap glucose as G6P
2) GLUT-2 requires high [glucose] gradient to pass glucose through plasma membrane. |
|
|
What does PFK1 do, how is it regulated
|
1) phosphorylation of fructose-6-phosphate (irreversible)
2) inhibited by ATP & citrate 3) activated by AMP |
|
|
How is the futile cycling of glycolysis and gluconeogenesis prevented
|
Key is dual enzyme PFK2/fructose bisphosphatase2 (PFK2 only active during high insulin levels)
1) Insulin -> +PFK2 produces F2,6BP -> +PFK1 & -F1,6BP(glycolysis) 2) Low insulin/high glucagon -> -PFK2, depletion of F2,6,BP -> -PFK1 & + F1,6BP(favouring gluconeogenesis) |
|
|
What are the 3 important fates of pyruvate
|
1) Produce Acetyl CoA (release CO2, produce NADH) -> lipogenesis or TCA
2) Produce oxaloacetate (acts as H atom acceptor from NADH, allowing passage into mitochondria OR first step of gluconeogenesis) 3) produce lactate in muscle under anaerobic conditions -> cori cycle -> liver |
|
|
Which of the LFTs is the most specific for acute liver damage
|
ALT
|
|
|
Which liver enzyme is also present in cardiac muscle, erythrocytes & skeletal muscle
|
AST
|
|
|
Which liver tests indicate cholestasis
|
ALP, GGT
|
|
|
Which type(s) of bilirubin are raised in acute hepatitis A (mechanism?, test?)
|
In acute inflammation, conjugated bilirubin is raised because it cannot be transported out of hepatocytes into biliary cannuliculi and is alternately shunted back to the bloodstream (urine shows bile)
|
|
|
List the steps that occur in the GI for breaking down proteins
|
1) stomach lumen: pepsin + HCl - > small peptides
2) trypsin, chymotrypsin break down into smaller peptides in lumen 3) carboxypeptidase is brush border enzyme -> individual AAs |
|
|
what are the enzymes that brreak down carbohydrates
|
1) pancreatic amylase; starch -> maltose
2) brush border: maltose -> glucose (maltase), lactose -> glucose + galactose (lactase), sucrose -> glucose + fructose (fructase) |
|
|
What is the chemical process of digestion and what is the energy requirement
|
Macromolecules are hydrolysed (no ATP requured)
|
|
|
Define asthma (3 parts)
|
1) bronchiolar hyper-responsiveness
2) airway inflammation 3) variable airway obstruction |
|
|
What 3 factors contribute to airway narrowing in asthma
|
1) thickening of smooth muscle layer
2) oedema 3) excessive mucous secretion with infiltrate of leukocytes |
|
|
List 3 types of asthma and their relationship with atopy
|
1) allergic asthma: early onset, other atopic symptoms (rhinitis, excema), raised IgE
2) intrinsic (later life, no atopy, no raised IgE) 3) occupational (chemical, dust or allergen induced) |
|
|
List the 5 types of atopy in encreasing severity
|
1) asymptomatic
2) allergic rhinitis 3) atopic dermatitis 4) asthma 5) anaphylaxis |
|
|
Describe the T-cell abnormality that occurs causing atopy
|
Th2 normally respond to parasite & helminth infestations but are activated by allergens.
|
|
|
Describe the 4 steps of airway inflammation in asthma
|
1) macrophage/dendritic cell presentation of allergen antigen to Th2 cells
2) Secretion of inflammatory cytokines by Th2 cells (IL4,5,9,13) 3) Migration of effector cells: early: mast and later: eosinophils and T-cells 4) inflammation, smooth muscle thickening, vascularisation |
|
|
After antigen presentation to naive Th cells, what factors are secreted and what cells are affected
|
1) Th0 -> Th2
2) Th2 secretes IL4,5,9,13 3) IL4 -> mast cells, IL5 -> eosinophils, IL 4,13 -> B cell maturation & proliferation -> IgE & mucus production, coating of mast cells with IgE |
|
|
What is the role of mast cells in asthma
|
IgE regognises allergen -> degranulation -> release of histamine (vasodilation & thickening of airways) and IF-a (smooth muscle contraction)
|
|
|
Describe the 2nd phase response of asthma
|
eosiniphils migrate and release leukotrienes (eg LTA4 -> receptors), macrrophages and T-cells -> mucosal swelling, inflammation) and TNF-a
|
|
|
What is the fundamental cause of asthma
|
Failure to develop tolerance during childhood
|
|
|
How can an allergy be cured
|
Immunotherapy: increasing low dose exposure (sc injection or SL drops) to allergen over period of months
|
|
|
What are the 2 interrelated abnormalities of asthma that are normally inherited
|
1) immunological deficits (atopy, inability to develop tolerance)
2) physiological abnormality: hyperresponsive airways |
|
|
List the common trigger factors of asthma
|
1) allergens: pollen, dust mites, pussy dander
2) pollutants: fags, pollution 3) occupational: isocyanates, welding smoke 4) other: exercise, cold air |
|
|
What 4 risk factors alert the possibility of asthma
|
1) family history of atopy
2) early sensitivity to allergens 3) infantile atopic excema 4) frequent infantile viral respiratory infections (rhino, rsv) |
|
|
What is one hypothesis explaining the rising level of asthma nad atopy in Western society
|
Hygeine hypothesis: less contact with far allergens and barn animals -> incomplete development of tolerance -> development of allergies & asthma
|
|
|
How much O2 is consumed at resting and exercise
|
1) 250mL
2) 3-4L |
|
|
Define total lung capacity
|
TLC = maximum inspiration - empty lungs or
TLC = vital capacity + reserve volume |
|
|
Define residual volume
|
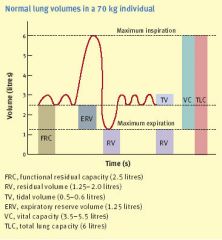
Volume remaining after forced expiration: alveolar + anatomical dead space
|
|
|
Define vital capacity
|
TLC (total lung capacity) - residual volume (think dead space)
|
|
|
What is the inspiratory reserve volume
|
After a normal breath in, the additional air it takes to fill the lungs (up to the TLC)
|
|
|
At the end of exhaling during tidal breathing, what are the two important reserves
|
1) Expiratory reserve volume: the amount we can further exhale by force
2) Functional reserve: the total volume remaining in the lungs ie the ERV + the RV (residual volume) |
|
|
Which volumes cannot be recorded by spirometry
|
Residual volume, therefore: functional residual capacity and total volume cannot be measured
|
|
|
Which muscles doe the medullary DRG (dorsal respiratory group) and the VRG (ventral group) control
|
DRG: (dindins) INspiration, therefore external intercostals and diaphragm
VR:: out: therefore internal intercostals |
|
|
How does the pons influence breathing
|
1) pneumotaxic centre inhibits medullary DRG to prevent overinflation and extended inspiration
2) apneustic centre: activates DRG but is overridden by pneumotaxic centre |
|
|
Which nerves control the main muscles of breathing
|
1) intercostal nerves
2) phrenic (diaphragm) |
|
|
describe the Hering-Breuer reflex
|
Stretch receptors in airways -> pneumotaxic centre in pons inhibiting the DRG in the medulla
|
|
|
What is the fate of glycerol attached to TAGs after they encounter a cell membrane
|
1) Lipoprotein lipase at the cell membrane -> glycerol + FFA
2) glycerol in plasma taken up by liver 3) forms DHAP |
|
|
What are the steps of fatty acid catabolism
|
1) Activation: FA -> FA-CoA in cytosol
2) carnitine shuttle: transport FA to mitichondrial matrix 2) B-Oxidation: FA-CoA -> many Ac-CoA (dehydrogenation -> FADH2, hydration, oxidation -> NADH, cleavage of Acetyl CoA from Acyl-CoA |
|
|
Describe the 3 stages of fatty acid oxidation and their location
|
1) B-oxidation: FA-CoA -> Ac-CoA in mitochondrial matrix NADH, FADH2
2) Ac-CoA -> TCA cycle (3NADH, FADH2 + GTP Co2) in mito matrix 3) Electon transport: NADH, FADH2 -> ATPs (uses O2, produces H2O) |
|
|
How are FFAs transported into the mitochondrion
|
1) If C1-14 then FA (NOT FA-COa) passes directly
2) If C16+ then FF-COa -> FF-COa-carnitine -> mitochondrial matrix |
|
|
What are the 4 products of 1 step of B-Oxidation
|
1) NADH
2) FADH2 3) CO2 4) FA-COa (2 carbons shorter) |
|
|
State the alveolar gas equation, what does it measure
|
PAO2 = 150-5/4(PaCO2) is a measure of the partial pressure of O2 in the alveloi
|
|
|
In types 1 & 2 respiratory failure, what do we expect regarding the ventilation perfusion mismatch
|
V/Q < 1
|
|
|
why does V/Q change at the apex of the lung ccompared to the base
|
1) less alveolar compliance, higher transmural pressure => V is lower
2) hydrostatic pressure on alveolar capillary bloodflow => Q much lower tnan fall in V => rise in V/Q |
|
|
When is O2 therapy effective in lung diseases (what types of diseases and respiratory failure situations)
|
1) Restrictive: all alveoli are inadequately oxygenated, V/Q < 1, therefore O2 therapy effective
2) Obstructive: O2 effective if alveoli partially blocked but not too many are completely blocked while others remain open. |
|
|
what are the features of type I respiratory failure
|
1) V/Q mismatch ie pA-aO2 > 10 because of increased shunting
2) pCO2 < 40mmHg due to hyperventilation |
|
|
What are 4 features of secretory epithelium
|
1) Polarized epithelium
2) Tight junctions 3) Ion channels on apical surface 4) Active transporters on the basolateral surface setting up ion gradients |
|
|
How is the chloride concentration gradient maintained in a chloride secretory cell
|
Na+/K+/2Cl- symporter at the basolateral membrane.
This is driven by secondary active transport via a Na+/K+ ATPase at the basolateral membrane |
|
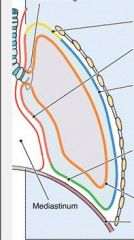
Name them
|
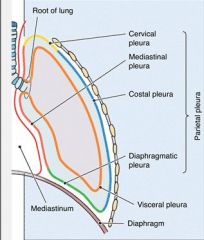
Nee to know the 3 parts of the parietal pleura
|
|
|
What is the cause of renal artery stenosis for younger and opder patients
|
1) fibromuscular dysplasia
2) atheroschlerotic |
|
|
List the 3 mechanisms of acid-base homeostatis
|
1) buffer system
2) respiratory compensation 3) renal compensation |
|
|
Describe how bicarbonate is reabsorbed in the proximal tubule
|

1) Bicarb in lumen is converted to CO2 + H2O by carbonic anhydrase
2) Na/H+ antiporter secretes H+ 3) CO2 + H2O diffuse thru epithelium to form H+ + HCO3- 4) NaHCO3 transported into interstitium by bicarb/Na+ symporter and bicarb/Cl- antiporter |
|
|
Describe how bicarb is produced de-novo in the distal tubules
|
1) CO2 + H2O -> H2CO3 in epithelium
2) H+ pumped into lumen by ATPase (primary luminal H+ ATPase) 3) HCO3-/Cl- exchanged in basolateral membrane |
|
|
Describe how bicarbonate is reabsorbed in the collecting ducts
|
1) CO2 + H20 forms bicarb and H+ in type A intercalated cells
2) H+ pumped into lumen by ATPase 3) H+/K+ exchanged by a 2nd ATPase in apical side 4) HCO3-/Cl- exchanged on basolateral side |
|
|
If a person is alkalotic, how do the renal tubules compensate
|
1) proximal tubules reabsorb less bicarb
2) Type B intercalated cells secrete bicarb and reabsorb H+ |
|
|
Describe the mechanisms whereby the kidneys compensate for metabolic acidosis
|
1) increased de-novo synthesis of bicarb by secretion of ammonium ion
3) phosphate and ammonium buffer excess H+ secreted into urine |
|
|
Describe the process of acute tubular necrosis caused by ischemia
|
1) depletion of ATP at thick ascending L of H and cessation of ion homeostasis, increased intracellular Ca++ -> cell death
2) production of oxygen free radicals -> cell membrane breakdown 3) severe reperfusion injury causing widespread tubular necrosis 4) debris and casts blocking tubular outflow |
|
|
Describe the 3 phases of acute kidney injury
|
1) Initiation: sudden fall in urine output, GFR & rise in creatinine & urea
2) Maintenance: several weeks with low urine output, increasing creatinine levels, urea and potassium. 3) Recovery: fall in creatinine, urea & potassium but abnormal urine production and electrolyte balance may persist |
|
|
List 5 causes of secondary HTN
|
1) RA stenosis
2) tumors (pheochromocytoma, aldosterone secreting) 3) pregnancy 4) cushings 5) coarctation of aorta |
|
|
What are the 9 (mnemonic) effects of ACE
|
Vasoconstriction, Aldosterone, Salt retention, SNS stimulation, ADH, Renin inhibition, Inotrope, Collagen deposition, Thirst
|
|
|
Clinical signs of hypertension
|
LVH
Retinopathy (I: tortuous arteries, II: A-V nicking, III: copper wire IV: haemorraging) Headaches Encephalopathy Acute renal failure |
|
|
Lifestyle modifications for hypertension
|
Salt
Fats Exercise Smoking & alcohol Weight loss |
|
|
Where do the 3 main types of diuretics work
|
1) Loop: NaKCl2 channel thick ascending LOH
2) Thiazides: NaCl symporter early distal (hypokalemia, hyperglycemia, hyperuricemia - weak acid) 3) Potassium sparing: blocks aldosterone receptors late distal, early collecting: decreased duct ENac channels (hyperkalemia) |
|
|
Describe rapid blood pressure controls
|
baroreceptors (carotid, aortic) -> vasoconstriction, HR, contractility via SNS
|
|
|
Describe the intermediate mechanisms for BP control
|
1) fluid compartment shift
2) RAS: vasoconstriction, Na retention, ADH, angiotensin, afferent arteriole constriction |
|
|
Describe how a decrease in blood volume triggers RAS
|
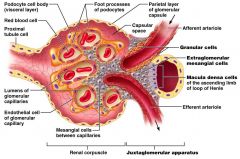
1) decreased GFR -> dilute distal tubular filtrate
2) Macula densa detects decreased Na 3) secretion of renin by granular (juxtaglomerular) cells |
|
|
List the 3 main areas angiotensin 2 acts on
|
1) brain: thirst, ADH
2) adrenal cortex: aldosterone 3) smooth muscle (arterioles) |
|
|
Why is the overall pressure within the glomerulus only 10mm Hg
|
1) hydrostatic pressure: +55
2) plasma colloid: -35 3) Bowman's capsule: -10 |
|
|
What is the formula for creatinine clearance
|
(U.V)/P where U= urine creatinine, V = urine volume (per minute), P = plasma creatinine
|
|
|
List the 4 rapid buffer systems in the bloodstream
|
1) bicrabonate
2) haemoglobin. 3) phosphate 4) proteins |
|
|
List the main intermediate buffer system
|
respiratory compensation
|
|
|
Describe how baroreceptors reduce BP
|
Carotid baroreceptors stretch -> glossopharangial nerve & aortic arch baroreceptors -> vagus nerve -> cardioregulatory centre in medulla -> increased para, decreased sympathetic tone -> decreased SR, SV and peripheral resistance.
|
|
|
Describe how chemoreceptors increase BP
|
Carotid chemoreceptors respons to decreased pH & O2, increased CO2-> decreased glossopharangial nerve tone & aortic arch chemoreceptors -> decreased vagus nerve tone -> medulla cardioregulatory centre -> decreased para, decreased sympathetic tone -> increased SR, SV and peripheral resistance.
|
|
|
List the medium to slow responses to a decrease in blood pressure
|
1) decreased ANP (vasodilation, sodium & water retention)
2) increased vasopressin (increased aquaporins and NaKCl transported in thick ascending LOH) 3) Decreased renal pressure diuresis -> renin secretion at macula densa by juxtaglomerular cells 4) RAAS |
|
|
Describe the temperature regulation mechanisms of the skin
|
1) vasodilation/constriction
2) sweat 3) arrector pili muscle (goosebumps) - decreases air flow around skin 4) insulation (fat in hypodermis) |
|
|
List the immune functions of the skin
|
1) barrier: keratinisation, desmosomes between cells (tight junctions), sebum & acidic sweat
2) Langerhan's cells (APCs that phagocytose) 3) SALT (IgA secretion) |
|
|
Describe the barrier roles of skin
|
1) dessication: overlapping cells and intercellular lipid
2) abrasion: keritanisation, cornification 3) UV: melanin from melanocytes in S.basale migrating to keratinocytes 4) bacteria, parasites: desmosomes, sebum & acidic sweat |
|
|
List the phases of acute tubular necrosis
|
Oliguric
Diuretic Resolution |
|
|
Define ejection fraction, what is "normal" and what cause it to fall
|
EF = SV/EDV x 100
Normal = 65-70% Low caused by cardiogenic shock, tamponade, aortic stenosis, coarctation of the aorta |
|
|
Describe what pulmonary wedge pressure measures
|
1) left atrial pressure (LAP=8-10mm if greater then LVF, if LAP=20mm then pul oedema)
2) pulmonary artery pressure (25/10mm) |
|
|
Describe the first steps when confronting an accident scene
|
D: danger, dial for ambulance
R: response: coma scale (EYES: open your eyes, VERBAL: can you hear me, what's your name, MOTOR: squeeze my hand) Airway: oropharyngeal tube Breathing: BVM (bag valve mask) Compression: 2/30 Defibrillation: right 2nd rib to left 10th rib |
|
|
Describe the effect of mitral stenosis on CO, MAP and compensatory mechanisms
|
1) ↓EDV ->↓SV-> ↓CO & MAP = COxPR, ↓MAP->↓ arterial volume & venous volume increases
2) uncompensated: pulmonary hypertension & oedema 3) compensations: LV hypertrophy |
|
|
Describe the effect of aortic stenosis on CO, MAP and compensatory mechanisms
|
1) ↓EF & SV=EFxEDV -> ↓SV & CO=SVxHR->↓CO & MAP=COxPr -> ↓MAP->↓ arterial volume & venous volume increases
2) uncompensated: pulmonary hypertension & oedema 3) compensations: LV hypertrophy |
|
|
explain the effects of sepsis on the cardiovascular system
|
1) distributive shock: fluid shift to intravascular compartment + vasodilation -> ↓blood volume & ↓PR & ↓EDV->↓SV->↓CO->↓MAP
2) compensation: aortic & carotid baroreceptors -> decreased tone of CN IX&X, decreased PNS & increased SNS from medullary cardiovascular centre -> ↑ HR, SV, PR |
|
|
A patient, following trauma, initiall presents with HR 120 & BP 110/65 but remains alert but sweaty, pale & thirsty. What is happening.
|
Compensated hypovelaemic shock: ie ↓EDV->↓SV->↓CO->↓MAP but compensatory mechanisms: aortic & carotid baroreceptors->↑tone of CN IX & X -> ↑SNS (sweat) & ↓PNS -> ↑HR, SV, PR (pallor)
Other mechanisms: endothelin (vasoconstriction), RAAS (vasoconstriction, decreased urine output, thirst) |
|
|
What are the biochemical consequences of von Gierke G6Pase deficiency
|
1) Glycogen -> G6P which cannot enter bloodstream -> hypoglycaemia
2) liver mass action effect - increased glycolysis -> lactic acidosis 3) accumulation of Acetyl CoA due to saturation of TCA cycle leads to lipogenesis (accumulation of triglycerides in liver) PLUS formation of ketone bodies -> ketoacidosis 4) hyperuricaemia due to pentose phosphate pathway -> accumulation of nucleic acid ribose molecules (purines) -> breakdown to uric acid |
|
|
What diet is necessary to treat von Gierke disease & why
|
1) uncooked cornstarch -> slow increase in blood glucose not triggering insulin and glycogen storage
|
|
|
Describe the short term adjustments to hypoglycaemia
|
1) SNS stimulation -> AD & NA -> tachycardia & CO, sweating, glycogenolysis in muscle
2) glucagon acts in liver & adipose: gluconeogenesis, lipolysis -> FFA (carried by VLDL) + glycerol |
|
|
Describe the long term adjustments to hypoglycaemia
|
1) formation of ketone bodies
2) increased blood glucose by GH, cortisol 3) activation of RAAS |
|
|
What is the role of the formation of lactate as an end product of glycolysis.
|
1) NADH forms from oxidation of glyceraldehyde-3-P to 1,3-bisphosphoglycerate
2) accumulation of NADH in anaerobic state inhibits conversion of pyruvate to Ac-CoA increasing production of lactate from reduction of pyruvate by NADH |
|
|
How and why are the kinetics of glucokinase and hexokinase different
|
1) Hexokinase: low Km (conc of glucose to reach 0.5Vmax -> high affinity) and low Vmax (easily saturable) allows equilibrium of liver & blood glucose conc. Also inhibited by acculmlation of end products - ATP and G6P
2) glucokinase: liver and B cells (high Km and Vmax) -> low affinity but not easily saturable -> requires high conc of blood glucose to trap G6P in B cells (to produce ATP to transport insulin vesicles) and hepatocytes following carbohydrate meal. |
|
|
How do different glucose transporters help maintain glucose homeostasis
|
1) GLUT 2: liver, B-cells & proximal tubules: low affinity (Km) hence requires high concentration of glucose to activate uptake.
2) SGLT1 in proximal tubule (glucose-Na symporter apical membrane 2nd active transport, GLUT2 basolateral) 3) SGLT1 intestinal epithelium 4) GLUT4 activated by insulin only |
|
|
PFK is a key regulatory enzyme in glycolysis. How is it activated & inhibited
|
1) inhibited by ATP, citrate (indicates saturation of TCA & oxphos) & low F2,6BP caused by inhibition of PFK2 due to high glucagon)
2) activated most strongly by F2,6BP (high insulin -> phosphorylated active PFK2 -> F2,6BP) |
|
|
What are the irreversible step enzymes in glycolysis and why are they regulated
|
1) hexokinase
2) PFK 3) pyruvate kinase because they are different to gluconeogenesis |
|
|
The first 2 steps of gluconeogenesis reverses the action of pyruvate kinase. What are they
|
1) carboxylation of pyruvate wiith CO2 by puruvate carboxylase to form oxaloacetate (also taken from TCA)
2) removal of CO2 from oxaloacetate to form PEP |
|
|
Briefly describe how frequency and volume are sensed by the inner ear
|
1) frequency: high frequency: proximal part of basilar membrane, distal is low frequencies - vibrates against hair cells
2) no of hair cells recruited determines volume |
|
|
Give 3 examples of conductive hearing loss
|
1) obstruction of auditory canal (cerumen, swelling of the canal, tumours)
2) impairment of tympanic membrane (perforation, glue ear or supprative OM) 3) impairmentr of the auditory ossicles (necrosis, otoschlerosis) |
|
|
Give 3 examples of sensoneural healing loss
|
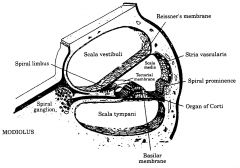
1) damage to the basilar membrane or hair cells by intense sound, ageing, frusemide or aminoglycosides)
2) damage to the cochlear nerve |
|
|
What forces are in balance to prevent alveolar collapse
|
1) forces promoting collapse: surface tension & elasticity
2) forces promoting opening: transmural pressure (diff between thoracic wall and alveolar recoil) + surfactant |
|
|
What is the transmural pressure relative to atmospheric pressure in 1) a normal lung and (2) a collapsed lung
|
1) normal lung about 4mm less than atmospheric pressure = alveolar pressure
2) pneumothorax: external pressure = alveolar pressure = transmural pressure |
|
|
Explain dynamic small airway closure
|
Forced expiration -> intrapleural pressure exceeds 1atm BUT alveolar pressure = 1atm - compression of bronchioles
|
|
|
Define physiologic dead space
|
Anatomical dead space (volume of airways) plus alveolar dead space (remaining air in alveolae after forced expiration)
|
|
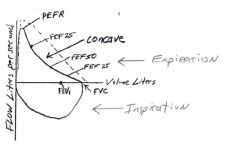
What does the flow volume loop spirometry indicate
|
Concavity indicates dynamic small airway obstruction in obstructive airway disease (asthma)
|
|
|
What happens to the heme from RBCs broken down in the spleen
|
1) macrophages convert heme to unconjugated bilirubin
2) transported by albumin to liver 3) conjugated to glucuronic acid 4) transported in bile to intestine 5) converted to urobilinogen by gut flora, some reabsorbed 6) Oxidised to brown stercobilins |
|
|
In prehepatic jaundice, what would you expect regarding diagnostic tests
|
1) ALT, AST, ALP normal
2) stools normal 3) urine normal 4) direct & indirect bilirubin elevated |
|
|
In hepatic jaundice, what would you expect regarding diagnostic tests
|
think intrahepatic cholestasis
1) ALT, AST, ALP elevated 2) stools normal 3) urine dark (bilirubin +++) 4) direct normal, indirect bilirubin elevated |
|
|
How are the 3 energy stores used to produce energy in the fasted state
|
1) Fats -> FFAs -> AcetylCoA (B-Ox) -> CO2 + H2O + energy
2) glycogen -> glucose -> pyruvate -> TCA -> ... 3) PRotein -> AAs -> glucose or ketone bodies |
|
|
Briefly describe the alanine cycle
|
muscle -> AAs -> glutamate + pyruvate -> a-ketoglutarate + alanine -> bloodstream to liver converted back to -> glutamate (to urea) + pyruvate
|
|
|
What is the Cori cycle
|
muscle: glycogen -> lactate -> liver: lactate -> gluconeogenesis -> glucose -> muscle
|
|
|
What happens to glucose metabolism (1) after a meal (2) between meals (3) starvation
|
1) liver: gluc -> glycogen + lipognesis (AcCoA -> FFA -> TAG -> VLDL) muscle & brain use glucose, heart uses FA
2) liver: glycogen -> glucose, B-ox of FFAs -> energy, muscle, heart use FFAs 3) liver: AAs -> glucose + ketone bodies, FFAs -> B-ox -> AcetylCoA -> ketone bodies (heart, brain insert into TCA cycle) |
|
|
Thinking diff diagnosis & anatomy, what structures could chest pain indicate
|
skin, fascia, costal nerves, pleura, lungs trachea, pericardium, heart, oesophagus, aorta
|
|
|
How would an ECG show an MI located at the inferior left ventricular myocardium
|
ST elevation would occur at the leads parallel to the depolarisation wave ie leads II, III and AVF
|
|
|
How is the cardiac axis determined
|
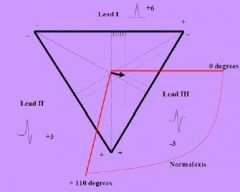
1) measure the net voltage of QRS at at least 2 leads
2) Mark on Einhoven's triangle 3) Normal is -30 to +110 degrees |
|
|
Describe the 4 phase action potential of myocytes
|
0) rapid Na+ channels (in)
1) Slow K+ (out) 2) Ca+(in)/K+(out-inactivated) 3) Rapid K+ channels (out) 4) Pacemaker potential: fast Na+ channels reopen at -55mv |
|
|
What happens to the myocite during ischemia
|
1) Resting membrane potential is reset higher due to decreased Na+/K+ ATPase activity.
2) Delayed after-depolarisation due to Ca++ increase due to Na+/Ca++ ATPase failure |
|
|
Explain in terms of Fick's Law why CO2 diffuses more readily across the alveolar wall than O2
|
Much greater diffusion coefficient ie
rate = AD/X(/\P) therefore small /\P is negated by very large D |
|
|
Given that arterial CO2=40mmHg, calculate alveolar O2
|
Alveolar gas equation is:
PAO2=PiO2-(PCO2/0.8) = 150-(40/0.8)=100mmHg |
|
|
Explain why severely anaemic patients cannot become cyanotic
|
Cyanosis requires around 50g/L deoxy Hb absolute. Severe anaemics do not have that concentration of haemoglobin.
|
|
|
How can a person have peripheral cyanosis without central cyanosis
|
Peripheral: local vasoconstriction -> deoxygenation of haemoglobin by local cellular respiration whereas arterial blood is normally oxygenated.
|
|
|
Explain why a TOF baby becomes cyanotic after exertion
|
1) Overriding aorta receives blood from both ventricles
2) Pulmonary artery stenosis increases RV afterload 3) Interventricular septal defect allows right to left shunt Overall: insufficient oxygenation of blood |
|
|
How is the type of respiratory failure determined clinically. Explain in terms of the type of shunts occurring.
|
Determined by response to O2:
Anatomical shunt: complete sealing of bronchioles (zero V/Q) unable to access O2. Other bronchioles with high V/Q and 100% oxygenated, O2 has little effect (except at bottom 1/2 of lungs with low anatomical V/Q). V/Q mismatch: where there is low V/Q, oxygen benefits. Where there is high V/Q (insufficient perfusion), O2 has no effect |
|
|
What is the main sign of type II respiratory failure and why
|
Ventilatory (pump) failure causes CO2 to rise. Can also be measured by respiratory acidosis
|
|
|
List the risk factors of developing CVD identified in the Framingham study
|
1) hypertension (> 140/90)
2) age 3) obesity 4) diabetes 5) LDL > 4.5mmol/L 6) gender (male) 7) genetic 8) lifestyle: smoking, physical inactivity, psychosocial (stress) |
|
|
The absolute risk of death from CVD of smokers is 343 per 1000 (ages 40-64). What does this mean?
|
Approximately 1 in 3 chance of dying of CVD before the age of 64
|
|
|
What types of arteries does atheroschlerosis affect. Name a few
|
1) medium - large (generally at bifurcations)
2) carotids, coronary, abdominal aorta (hear bruits), femoral, popliteal |
|
|
Describe the differences between the two main types of arteries
|
1) elastic: many layers of elastic lamella separating smooth muscle cells within the medial layer
2) muscular: two layers of elastic lamella separating the medial thick muscular layer from the tunica adventitia and tunica intima |
|
|
What steps are involved in the initiation of an atheroschlerotic plaque
|
1) accululation of extracellular lipid
2) sequestration of low density lipoproteins within the tunica intima 3) oxidation of lipoproteins -> inflammation |
|
|
Describe the process of recruitment of leukocytes in atheroma formation
|
1) expression of adhesion receptors (selectins & adhesins)
2) rolling adhesion + fixed adhesion to endothelium 3) diapedesis of neutrophils into tunica intima 4) secretion of cytokines attracts more inflammatory cells (mainly monocytes) |
|
|
Once lipoprotein has been sequestered into the tunica intima and inflammation has begun, describe the next steps
|
1) monocytes mature into macrophages and proliferate (CSF1)
2) macrophages try to mop up excess lipioprotein, forming unstable foam cells containing oxidised lipoproteins. 3) inflammation continues: PDG: proliferation of smooth muscle in tunica intima, organisation process where fibrotic remodelling occurs to expand vessel size (collegen, proteoglycans) & calcification occurs 4) lumen decreases (swelled with lipoprotein & foam cells)as vessel size reaches limit 5) endoendothelial rupture -> formation of thrombus 6) formation of aortic anaeurism -> rupture |
|
|
What are the 3 hypotheses of atheroma formation
|
Responses to:
1) injury 2) lipid deposition 3) oxidative modification (of lipoproteins) |
|
|
Describe the response to injury hypothesis of atheroma formation
|
1) injury to endothelial cells (lipids, smoking, hypertension)
2) expression of selectins, adhesins etc & attraction of neutrophils 3) "leakage" of LDL into tunica intima -> foam cell formation, chronic inflammation, retention of lipids by binding to proteoglycans (part of organisation/repair process) |
|
|
Descrribe the response to retention hypothesis of atheroma formation
|
1) hyperlipidemia and possible increased permeability caused by hypertension etc. allows migration of lipoproteins into tunica intima
2) retention of lipoproteins by proteoglycans -> chronic inflammation and atheroma formation |
|
|
Describe the oxidative modification hypothesis of atheroma formation
|
1) LDL migrates into lesion prone sites (eg at bifurcations) but not immediately antigenic
2) LDL subjected to oxidation by resident cells 3) oxidised LDL now antigenic and target of macrophages -> foam cells -> fibrosis & remodelling -> formation of atheroschlerotic plaque |
|
|
List the steps of fat & cholesterol digestion & transport
|
1) emulsified fats broken down to FFA's and reassembled into TAGS & packaged into chylomicrons by intestinal mucosa
2) transporrt via lymphatics to bloodstream. Lipoprotein lipase on endothelium, activated by APO-C2 on chylo, releases FFAs & glycerol 3) FFAs taken up by cells |
|
|
List the steps of VLDL production & its role
|
1) apoB-100 formed in liver
2) extracellularly add APOE & C2, from HDL (recycled from chylomicron) 3) VLDL transports TAGs to tissues |
|
|
Describe the formation of LDL & its role. What disorder causes its persistence in the blood
|
1) VLDL releases TAGs
2) returns apoC-2 to HDL, retains APO100 and carries cholesterol only to tissues 3) encounters LDL receptor and is phagocytosed. If receptor is absent then familial hypercholesterolemia |
|
|
What cycle of events causes high LDL/VLDL in familial hypercholesterolemia
|
1) liver cells unable to endocytose LDL and IDL (receptors absent or abnormal)
2) liver cells respond to apparent "shortage" by synthesizing cholesterol 3) cholesterol further increased by release from liver via VLDLs |
|
|
Explain the role of HDLs and why they are "good" cholesterol
|
1) receivers and donors of apoproteins (apoE, apoC2) to allow conversion between the different lipoproteins
2) store of extrahepatic cholesterol via "trapping" of cholesteryl esters 3) esters released only by donation to VLDL -> LDL -> endocytosis, or HDL/cholesteryl metabolised in liver |
|
|
What is the main role of ApoB-100?
What is its role in atheroschlerosis? |
1) binding to LDL receptors allowing endocytosis of LDL & cholesteryl esters
2) LDL containing oxidised cholesteryl esters is endocytosed and broken down by macrophages. Oxidised cholesteryl esters is retained by macrophages -> foam cells -> inflammation & fibrosis. |
|
|
What pathological outcomes can occur with tubular obstruction
|
1) inadequate perfusion & necrosis
2) infection 3) upstream hypertrophy due to pressure,, downstream atrophy, atrophy of surrounding tissues due to hypertrophy of tubes eg hydrocephaly or consequences from loss of blood flow (eg stroke) 4) retention of secretions, formation of stones |
|
|
What is the most common mutation causing CF
|
DeltaF508 (delta = deletion, F = phenylalanine)
|
|
|
CF patients often sufferr from bronchiectasis. Define it.
|
chronic necrotising infection of the cronchioles & bronchi causing abnormal dilatation
|
|
|
Describe the action of insulin at its receptor
|
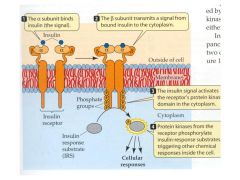
1) binding to alpha domain
2) autophosphorylation of protein kinases 3) phosphorylation of insulun response substrate (IRS) triggering phosporylation of other enzymes. |
|
|
List the significant energy producing steps of the TCA cycle
|
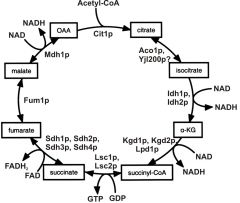
1) isocitrate (5C chain with OH) oxidised to keto acid + NADH
2) alpha-kg (5C) undergoes oxidative decarboxylation to form succinate (symmetrical 4C di-acid) forms GTP & NADH 3) oxidative dehydrogenation of the central C-C bond forming C=C (fumarate) and FADH2 4) after hydration of fumarate to malate, the C-OH is oxidised to C=O forming ocxaloacetate and NADH 5) total energy = 3 NADH, FADH2, and GTP |
|
|
Describe the process of cardiac muscle contraction (ie actin, myosin etc)
|
1) myosin heat primed by ATP
2) Ca++ bound to troponin shifts allows exposure of actin binding site 3) myosin attaches to binding site on actin and "pulls" 4) Ca++, released. actin site covered, myosin returns to "cocked" state with ATP |
|
|
Explain how acetyl-CoA from pyruvate becomes available again for lipogenesis
|
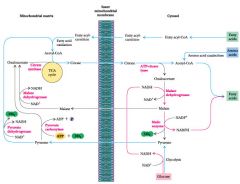
1) pyruvate freely enters mitochondrial matrix
2) pyruvate -> acetylCoA + NADH in mitochondria 3) acetyl-CoA hops on citrate/malate shuttle ie AcCoA + oxaloacetate -> citrate -> cytosol -> oxaloacetate + acetylCoa 3) oxaloacetate + NADH -> malate -> mitochondra OR lipogenesis |
|
|
List 4 properties of an ideal plasma marker (eg for cardiac or liver damage)
|
1) plasma non-specific but highly tissue specific
2) measurable (easily and cheaply) 3) sensitive test (rapidly released, long lasting) |
|
|
List 4 areas in the myocardium capable of releasing specific plasma markers
|
1 and 2) nucleus and mitochondria - CK-MB
3) myoglobin 4) myocytes (troponin) |
|
|
Why is lactate dehydrogenase not a great marker for an MI. How can this be overcome?
|
1) number of isoforms exist normally in plasma ie LD2 in WBC
2) compare levels of isoforms LD1 (cardiac) to LD2(WBC). If LD1 > LD2 then probable MI |
|
|
Why is creatine kinase not a great marker for an MI. How can this be overcome?
|
1) number of isoforms exist normally in plasma ie CKMM in muscle and some CKMB in myocytes
2) compare levels of isoforms CKMB (cardiac) to total creatine kinase. If CKMB > 3%-6% total serum creatinine kinase then probable MI |
|
|
Describe how damaged venous endothelium triggers coagulation. How can a crucial step be stopped by heparin
|
1) endothelium releases tissue factor
2) factors VII, IX then X activated 3) factor Xa produces thrombin which converts fibrinogen to fibrin 4) antithrombin III is activated by heparinn, preventing thrombin formation |
|
|
Describe how damaged arterial endothelium triggers coagulation
|
1) platelets adhere to von willerbrand's factor & collegen
2) thromboxane a2 -> platelet cross linking |
|
|
How is a venous thrombus eventually broken down
|
1) tissue plasmin activator converts plasminogen to plasmin
2) plasmin breaks down fibrin |
|
|
Draw the cardiac cycle showing ECG, LV pressure and volume, LA pressure
|
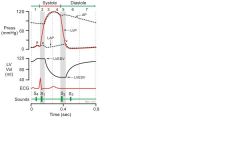
|
|
|
Explain how pulsus paradoxus arises during acute asthma
|
1) use of accessory inspiratory muscles (sternocleidmastoid, scalene, pec minor/major) causes high transpulmonary pressure (PA - Ppleural).
2) The -ve pressure causes capacitance of blood in lungs (high vascular compliance) 3) reduced venous return and systolic BP |
|
|
Why doesn't a complete anatomica shunt occur during acute asthma
|
1) hypoxic vasoconstriction
2) collateral air passages |
|
|
Give 2 examples of conditions causing type I respiratory failure
|
1) V/Q mismatch: asthma, pul embolism, COAD, consolidation
2) Impaired diffusion: pulmonary oedema |
|
|
Give 3 examples of conditions causing type II respiratory failure
|
1) upper airway obstruction
2) respiratory centre depression (opioids) 3) fatigue: severe presentations of asthma, COAD |
|
|
Define functional residual capacity and what is it a measure of
|
FRC = ERV + RV
Measure of air remaining in lungs at the end of passive breathing. Increases in emphysema because of decrease in elastic recoil, decreases in restrictive lung disease |
|
|
Explain why VC is reduced in obstructive airway disease
|
Inability to expel air completely due to dynamic small airway closure. Increased FRC (functional residual capacity), hence decreased vital capacity
|
|
|
Explain why VC is reduced in restrictive airway disease
|
Reduced inspiratory reserve volume results in reduced vital capacity
|
|
|
List all the factors contributing to small airway closure in asthma
|
1) hyperresponsiveness to triggers (allergens, exercise, chemicals eg aspirin/NSAIDs)
2) inflammation: oedema, vascular dilation, mucus secretion 3) chronic inflammation airway remodelling: smooth muscle hypertrophy |
|
|
Describe the lates stage immunological steps in developing asthma
|
1) APC presents antigen to Th2 cell
2) Th2 cells stimulate proliferation of B cells producing IgE 3) Activated Th2 secrete cytokines attracting eosinophils 4) Eosinophils activated by IgE -> histamine, heparin, TNF-a -> oedema, vascular permeabuility to other lymphocytes, smooth muscle proliferation |
|
|
Which drugs are appropriate for early stage asthma
|
Bronchospasm caused by mast cells releasing CysLT (leukotriene), therefore: B2 agonists, theophylline, leukotriene receptor antagonists, mast cell stabilizers
|
|
|
Which drugs are appropriate for exacerbations of late stage asthma
|
Need to inhibit recruitment and proliferation of Th2 cells and proliferation of B-cells producing IgE, proliferation of eosinophils
|
|
|
Describe the cause of referred pain during MI
|
Pain is referred to the sympathetic chain and dorsal roots T1-T4 which include dermatomes for the neck, jaw, shoulders, arms and stomach
|
|
|
Explain why pink, frothy sputum, hyoxia and tachypnoea may be produced during an MI
|
Reduced LV ejection fraction due to ischemia -> increased left atrial pressure -> increased pessure in the pulmonary circulation -> oedema
|
|
|
Which ECG leads refer to which region of the heart
|

See diagram
|
|
|
What two forces acting on the lungs oppose each other when breathing
|
1) lung recoil (always acts to colllapse the lung)
2) intrapleural pressure (always negative and acts to expand the lung) |
|
|
What can we say about intrapleural pressure and lung recoil when the lung volume is the functional residual capacity
|
They must be equal as this follows a normal expiration of the tidal volumegiving FRC.
|
|
|
What two classes of material are not freely filtered by the kidneys. How are they changed to be freely filtered.
|
1) proteins -> AAs
2) fats - conjugated by glucuronidase enzymes in liver |
|
|
What is the formula for inulin clearance given the concentralions of inulin in a volume of urine as well as the plasma conc
|
note that the amount of inulin cleared, q = U x Cu = V x Cp and V (the plasma volume) = clearance
|