Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
36 Cards in this Set
- Front
- Back
|
Definition of Pharmacology, Drugs
|
Pharm: the science of drugs, prep, uses, effects
Drugs: any chemical agent that has an effect of processes associtated with life |
|
|
Receptor Characteristics
|
1. Must be biologically important-proteins
2.Structural feature which permit drug specificity 3. Drug binding site and biologically active site(can be same, often seperate) |
|
|
Molecules capable of serving as receptors
|
1.Enzymes: complex, impt, catalytic site
2. Membrane Proteins: most important, EC triggers something intracellularly 3. Nucleic Acids: can be targets, set up rxn cascades 4. Polysaccharrides: On cell membrane, involved in cell interactions |
|
|
Major classes of signal transduction receptors(all membrane bound except intracellular receptors), examples and structures
|
1. Ligand Gated-5 polypeptides with alpha helices in MB EX: Nicotinic Receptor
AcH binds Receptor(2 of them)-->opens channel, Na rushes in-->contraction 2. G-protein-linked Receptor-MB bound 7 polypeptides, Ex. Epinephrine, Rhodopsin, cascade of intracellular events 3. Catalytic Receptor: single polypeptide, drug binding site and catalytis site. Ex. Oncogenes, come in pairs 4. Intracellular- binds LIPID soluble drugs. EX: Steroid Receptors-steroid binds receptor which migrates to nucleus and binds dna-->alters transcription |
|
|
Factors which affect Drug Binding Forces
|
1. bond envergy
2. bond number 3. attractive distance 4. Bond requirements |
|
|
Bond Force Types:
|
1. Covalent-Very strong hence not used in drugs much. Ex. Asprin
2. Ionic Bond-medium strength, varies with distance of bond(squared). 1st type of attraction, non specific 3. Hydrogen-weak, short distance but can have multiple bonds and provides some specificity(FOHN). Because multiple can be strong 4. Cationic pi bond-+ chaged molecule with ammonium group interacts with benzene ring's e- cloud. similar to ionic but weaker(like hydrogen) 5. Van der Waals-weakest and short distance but provides most specificity. Molecular match can lead to collective strength in bonds 6. Hydrophobic: helps stabilize bonds. Attraction of H20 to itself forces drug to receptor site |
|
|
Cooperation of binding forces example:
|
Epinephrine and B-adrenergic R
B-adrenergic R: 7-G protein linked Initial attraction: aspartic acid is negative charge on receptor, attracts + charge group of epi Orientation: hydroxyl groups on benzene of epi form H-bonds with serine resides(receptors) Strengthening: Vdw-->binding binding-->g protein cascade-->heart beat stronger |
|
|
Consequences of Drug Binding
|
1. Conformational Induction-changes confirmation of receptor to either bind or not bind drug
Ex. Allosteric inhibition(epi) or activation 2. Conformational selection- Receptor exists in 2 phases, passive or active. Drug binding to each phase further stabilizes the phase. Ex. indirect allosteric, Active-drug binds active Rs-->shifts eq so, body makes more active Rs Inactive-drugs binds inactive Rs, shifts eq so active Rs become inactive 3. Effector site blocking actions-drug competes with substrate at catalytis site, either does the same thing as substrate or blocks its activity. Ex. Carbonic anhydrase: forms CO2, H2O which causes altitiude sickness, inhibit/prevent sickness by blocking catalytic site |
|
|
Conformational Diagram Constants
|
Kia- Eq between active/inactive R
Kdi-Eq b/w Ri + D/ RiD Kda- eq b/w Ra + D / RaD KDia- eq b/w RaD/ RiD |
|
|
if R preferentially binds Ra how does the Eq shift in conformational selection?
|
Ri --> Ra
|
|
|
Structure Activity Relationships:
|
-Small changes in molecular structure can cause large change in drug action
-because Rs are so conserved and similar in structure. Modding drug leads to new binding to new receptor. Ex. prozine, antihistamine-->schizo |
|
|
Dose-Response Relationships Goal
|
dose to effective yet not enough to be toxic
|
|
|
Dose-Response Relationships: Occupational Theory
|
Single drug binds to a single receptor to give singular effect
Michaelis Mention Equation: D+R <-> DR <-> effect K1= binding K2=dissociation Kd= K2/K1 = dosage of drug(D) as which Contraction is 50% Effect = Max effect x D/ Kd + D Drug dose = X axis Contraction percentage = Y-axis |
|
|
Potency
|
Amount of drug needed to attain a desired effect.
More Potency = less drug, lower dissociation constant=likes to bind to R |
|
|
Efficacy
|
Max effect one can achieve with a given amount of drug
|
|
|
Graph of Contraction vs. log dose:
Threshold dose Ceiling effect Linear component |
threshold: below which there is no effect
ceiling effect: beyond which you get no effect Linear component-therapeutic range, constant predictor of effect/dose |
|
|
Agonist
|
Any drug that creates a change +/-
Relative Effect = a x D / Kd + D Effect = e x Rt(receptors) x D / Kd + D Full: produce ceiling effects, maximum effectiveness but K3(intrinsic activity) may differ(potency). Max intrinisc activity(a =1) but may differ in affinity Partial: produce submaximal effects regardless of amt of drug given. Less intrinsic activity(0<a<1), may also differ in potency as well as affinitiy |
|
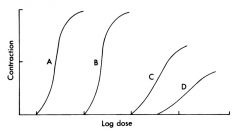
A vs. B
C vs. D AB vs. CD |
AB: Potency
CD: Potency, Efficacy and intrinsic activity ABvsCD:potency, efficacy, intrinsic activity |
|
|
Antagonists Definition
|
Block action of other drugs, only observed in presence of agnost
Ex. Atropene blocks action of ACH |
|
|
Antagonists Types Classic
|
competitive, decrease agonist potency
-->need more drug to overcome anatgonist but can still achieve max efficacy |
|
|
Antagonists Types Partial
|
act like competitve antagonist with full agonist, partial can block full agonist as it occupies receptors in the same system
|
|
|
Competitve Antagnoists
|
shifts curve to right
1. Agonist has less affinity for R due to competition by other drug 2. Intrinsic activity unchanged-can achieve max effect with more agonist 3. increase competitve antagonist-->curve keeps shifting to right |
|
|
Noncompetitive Antagonist
|
Curve shifts to right, shortens, maximum effect reduced, pushes curve down
1. Small Potency change 2. Changes intrinsic activity 3. Reduces effect but dose for 1/2(KD) stays the same -->same as reducing number of receptors similar to if you removed some receptors from the system, binds to site other than the one used by the agonist cuasing a chang ein conformation preventing binding adding more agonist does not overcome, does NOT change binidng affinity or Potency |
|
|
Allosteric Modulators
|
1. Alter Kd -->competitve antagonist
Look like competitive antagonist but are limited in how much they can shift to the right 2. Alter intrinsic efficacy-->noncompetitive BUT reversible, true noncomp. is not reversible -can do both, as well as INCREASE agonist binding/efficacy |
|
|
Failure of Occupation Theory
|
1. 1 drug reversible binds single R
F: Asprin binds irreversbly, nictoinic R needs 2 ach to bind 2. Drug binding indp. of other drug recep interactions-usually true but not always 3. All receptors are identical and equally accesible-rarely true F: 1. beta R different in each person 2. Desensitization; R lose ability to bind well due to conformaitonal changes 3. some Rs maybe be covered by proteins, lipids etc. 4. Only small portion of drug is bound-true almost always 5. Response is proportional to receptor binding and is time independent-false time dependent F: 1. Receptor access-bodies have redundancy. don't need to stim all receptors to get max effect |
|
|
Spare Receptor theory
|
1. More Rs available than needed to make max response
2. Agonist only has to bind portion of Rs for full effect(10%) 3.Competitve antagonist must block large fraction of Rs to have strong effect.(acetylzolamide needs to be 99.85 blocked to get 1/2 block of carbonic anhydrase activity) 4. Non-competitive antagonist may look like competitive antagonist -dependent on effect. For example if agonist only needs 10% to for full effect, and a non-competitive antagonist binds 50% of them, you can still get max effect by simply adding more agonist Explains why this may look like a compettive antag at first-->no way to tell b/w non-comp/comp unless given full information. If effect can be overcome assume competitive unless curve changes. |
|
|
Stimulus Coupling System
|
F(tissue coupling) x e x Rt / Kd + D
efficient: don't need lots of ions to get effect inefficient: need lots of ions to get effect |
|
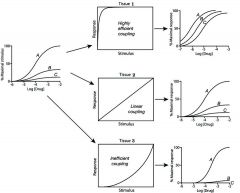
Explain
|
High efficient- dont need much to create effect thus not as many receptors needed to get max response-->all 3 drugs look like full agonist
linear coupling-increase in stim directly proportional to increase in response seen(occupation theory) Inefficient-needs high stimulus for max response. A-potency drops, but can still produce max response B-decrease from original stimulus c-cannot produce any response as stimulus is sub-threshold, looks like a comp. antagonist as it competes for the R Drug can be agonist/antagonist depending on system |
|
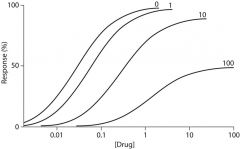
graph of what?
|
spare receptors and noncompetitive antgonism
|
|
|
Inverse Agonist
|
block effect of agonish and has opposite effect of agonist
most of the time Rs inactive w/o presence of ligand-->may be more inverse agonist that we know |
|
|
Antagonist + agonist(full/partial)
|
Antagonist blocks effect of agonist, Ra/Ri increases
|
|
|
Agonist + Partial Agonist
|
Low-increase total effect
high-competes for receptor sites, reducing total effect of full agonist Ex. narcotic analgesia small-analgesia, large - partial agonist blocks effects of full agonist |
|
|
Antagonist alone
|
-Fixes Ra/Ri ratio, block activity of both agonist and inverse agonist
|
|
|
Anatgonist + inverse against
|
Antagonist blocks effect of inverse agonist, Ri increase relative to Ra
Ri increases relative to Ra thus see an effect of absence of agonist and effect of no R ex. valium |
|
|
Receptor Indepedent drug rxns
|
chemically reactive agents
physically active drugs Counterfeit biocehmical consituents |
|
|
chemically reactive agents
Still have potency/dose issues! |
-no receptor
ex: disinfectants, alkylating anticancer drugs physically active: colligative properties, physical presence occupies a space not dependent on chemical properties Ex. Mannitol-changes in oncotic pressure in blood cause stuff to re-enter blood steam, draws fluid from brain cells removing excess waste H2O2-bubbles remove debris and killing chemically Counterfeit biochemical constiutents -5 bromouracil-looks like thymidine when incorporated into DNA, weakens or kills cells. |