Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
235 Cards in this Set
- Front
- Back
|
Pharmacodynamics
|
is the study of the biologic activity that a drug has on a living system. It includes a study of the mechanisms of action of the drug and the exact processes that are affected by it.
|
|
|
Pharmacokinetics
|
deals with the magnitude and time course of drug effect, and it attempts to explain these aspects of drug action through a consideration of dosage and the absorption, distribution, and fate of chemicals in living systems.
|
|
|
Bioavailabitity
|
indicate the fractional extent to which a dose of drug reaches its site of action (how much blood is available in the blood for the receptors)
|
|
|
Enteral
|
oral, sublingual, rectal
|
|
|
Parenteral
|
o Intravenous, Subcutaneous,
o Intramuscular, Intra-Arterial, Intrathecal |
|
|
Topical
|
o Transdermal, Mucous membranes, Eye, Ear
|
|
|
Advantages of Oral Ingestion
|
o Safest, most convenient, most economical
o Daily administrations and / or for a long period of time o Auto-administration |
|
|
Which is safest way of administering a drug?
|
Enteral --> ORALLY
|
|
|
Disadvantages of Oral Ingestion
|
- Disadvantages
o Limited absorption - physical characteristics o Emesis - irritation of the GI mucosa o Destruction - digestive enzymes or low gastric pH o Presence of food and other drugs o Cooperation on the part of the patient o Metabolization - Enzymes of the intestinal flora, mucosa, or liver o First-pass metabolism |
|
|
First-Pass Metabolism
|
lose a lot in hepatic portal system BEFORE the drug is even able to find the receptor --> esp. pts. with liver problems
|
|
|
Advantages of Rectal Ingestion
|
o less hepatic first-pass metabolism (50% less)
o do it when patient is unconscious or vomiting |
|
|
Disadvantages of Rectal Ingestion
|
o Absorption - irregular and incomplete
o Irritation of the rectal mucosa o Diarrhea |
|
|
Which method has the FASTEST ABSORPTION?
|
Parenteral - Intramuscular
|
|
|
Advantages of Parental Intramuscular Absorption
|
o Rapid absorption (FASTEST ABSORPTION)
o Possible Slow, constant absorption -by repository vehicles |
|
|
Disadvantages of Parental Intramuscular Absorption
|
o Big volumes
o Irritations, accidents o Paraesthesia, accidental intravenous o Cost; Trained people |
|
|
Advantages of Parental Intervenous Absorption
|
o Bioavailability - complete and rapid (100% of the drug reaches the blood !!!)
o Avoid absorption (no absorption… as you are directly placing drug into the blood) o Drug delivery - controlled, accurate and immediate o Big volumes; irritating solutions |
|
|
In which method is there NO ABSORPTION?
|
Parental Intravenous --> you are placing it directly into the blood
|
|
|
Advantages of Inhalation
|
o Almost instantaneous absorption
o Avoidance of hepatic first-pass loss o Pulmonary disease - local application |
|
|
Disadvantages of Inhalation
|
o Local and systemic reactions to allergens
|
|
|
Advantages of Topical Application
|
o Rapid absorbance in mucosa
o Local and systemic effects |
|
|
Disadvantages of Topical Application
|
o Intact skin - lipid barrier
o Dependent on the blood flow o Unwanted systemic side effects |
|
|
The cell membrane is a bilayer of amphipathic lipids with their hydrocarbon chains oriented inward to the center of the bilayer to form a continuous _______ phase and theirs _______heads oriented outward
|
hydrophobic heads inward
hydrophillic heads outward Therefore.... in order to reach, you must cross the hydrophillic heads... but to leave the cell membrane, need to encounter hydrophobic heads |
|
|
Oil/water partition coefficient
|
- drug ususally has a # that will tell you whether it is more lipophilic or hydrophilic
- “1” means there is an equal amount of hydrophilic and hydrophobic which is IDEAL so it can cross - A HIGH coefficient means it is lipophilic so it is harder for the drug to reach the cell membrane … HOWEVER , it is easier to cross the membrane - Lipophilic drugs are good drugs to cross the blood-brain barrier |
|
|
Passive diffusion
|
- Concentration gradient across the membrane
- Lipid-water partition coefficient of the drug - Surface area exposed to the drug |
|
|
Active transport
|
- Requirement for energy
- Movement against an electrochemical gradient - Saturability, selectivity, and competitive inhibition by co-transported compounds |
|
|
Facilitated diffusion
|
Carrier-mediated transport process in which there is no input of energy
|
|
|
What form of the drug will be able to cross the barrier?
|
ONLY the non-charged form
|
|
|
Will an acidic drug be absorbed faster in the stomach (acidic) or intestines (basic) ?
|
Stomach
**** However…. The time in the stomach will be shorter b/c NOT meant to absorb… just meant to breakdown. Therefore…. RATE is higher in the stomach, but MORE is absorbed in the intestines, just much slower for acidic drugs |
|
|
Distribution of a Drug is Dependent Upon
|
- Drug’s physicochemical properties
- Cardiac output and regional blood flow - Capillary permeability - Tissue volume |
|
|
Where are most basic drugs trapped?
|
in the STOMACH
--> even stuff like cocaine, even if inhaled |
|
|
Plasma Proteins
|
Some drugs .. For example… have like 30% bound to plasma protein (especially albumin) … so the other 70% is free in the blood
So the plasma protein can be considered a reservoir - This binding is capacity limited because the number of binding sites is finite. - Drugs differ tremendously in their affinity for plasma proteins; the percentage of binding of individual agents ranges from 0% to 100%. |
|
|
How would you transfer something across the blood-brain barrier?
|
- Most common way of transport in CNS is via transporters
- Higher the lipophilic… higher the chance to reach the CNS - Meningeal and encephalic inflammation increase permeability |
|
|
Placental Transfer of Drugs
|
lipid-soluble drugs pass thru the placenta
--> but fetus exposed to ALL drugs taken by mother |
|
|
What types of compounds are most easily removed from the body ?
|
- Polar compounds will not be reabsorbed…. Will exit the body
- Lipid soluble NEED to be metabolized or will go back |
|
|
A major factor influencing the reabsorption of weak electrolytes from renal tubular fluid is:
|
the pH
--> Depending on the rate of H+ secretion, the urinary pH may vary from 4.5 to 8.0. |
|
|
Weak acids such as aspirin and phenobarbital are reabsorbed more effectively under _________; the reverse is true for weak bases such as amphetamine and ephedrine.
|
acidic conditions
|
|
|
A common strategy in the face of aspirin toxicity is....
|
A common strategy in the face of aspirin toxicity is to promote salicylate elimination through alkalization of the urine by the systemic administration of sodium bicarbonate.
|
|
|
Where do we want the drug.. intracellular or extracellular?
|
Intracellular b/c we wanna absorb the drug
|
|
|
When we want to eliminate… where do we want the drug?
|
Extracellular so it can go off in the urine
|
|
|
Where are acidic drugs eliminated?
|
In the BASIC urine
|
|
|
Biliary Excretion
|
AKA "fecal" excretion
-Substances excreted in the feces -Un-absorbed orally ingested drugs -Drug metabolites excreted in the bile -Drug metabolites secreted directly into the intestinal tract and non-reabsorbed -Substances with high molecular weight (higher than 500) |
|
|
Enterohepatic recycling
|
Reabsorption of molecules excreted through the bile can occur, such as with the laxative phenolphthalein.
Such enterohepatic recycling can prolong the duration of action and may continue ad infinitum until the system is interrupted (e.g., by metabolism, curtailment of bile flow, or ingestion of a drug chelator). |
|
|
First-pass metabolism
|
The first-pass effect (also known as first-pass metabolism or presystemic metabolism) is a phenomenon of drug metabolism whereby the concentration of a drug is greatly reduced before it reaches the systemic circulation. It is the fraction of lost drug during the process of absorption which is generally related to the liver and gut wall.
Notable drugs that experience a significant first-pass effect are imipramine, morphine, propranolol, buprenorphine, diazepam, midazolam, demerol, cimetidine, and lidocaine. |
|
|
Which type of drug receptor is irreversible?
|
Covalent
|
|
|
Affinity
|
Degree of attraction between the drug and its receptor
|
|
|
Potency
|
Amount of drug (mg) needed to produce an effect
--> Potency is usually a matter of little importance because a drug that is very potent regarding its desirable effects is often equally potent regarding undesirable effects. In the intact animal (or patient), potency is influenced by the drug's ability to reach the receptor (determined by the rate of absorption and the patterns of distribution and elimination) in addition to the agent's intrinsic activity and receptor affinity. |
|
|
Intrinsic Activity
|
The ability to produce an effect (once the drug is bound to its receptor)
-Has nothing to do with affinity -Basically… whether or not there is any effect when bound to the receptor (i.e. Beta Blockers has no intrinsic activity) |
|
|
Examples of Cytoplasmic 2nd Messengers
|
cyclic AMP, cyclic GMP, cyclic ADP-ribose, Ca2+, inositol phosphates, diacylglycerol, nitric oxide.
|
|
|
Cytoplasmic 2nd Messengers
|
- Enzyme or transport protein that creates, moves, or degrades a small metabolite or ion
- They can diffuse in the proximity of their binding sites and convey information to a variety of targets, which can respond simultaneously to the output of a single receptor binding a single agonist molecule |
|
|
G-Proteins
|
G proteins are heterotrimers consisting of α, β, and γ subunits. After receptor activation, guanosine diphosphate attached to the α subunit is replaced by guanosine triphosphate, and the heterotrimer splits into the α monomer and βγ dimer.
Many, but not all, of the observed cellular actions are caused by the α |
|
|
Agonists
|
Bind to physiological receptors and mimic the regulatory effects of the endogenous signaling compounds
|
|
|
Antagonists
|
Bind to receptors without regulatory effect, but their binding blocks the binding of the endogenous agonist
(Antagonist is NOT THE OPPOSITE but rather, blocks) |
|
|
Partial Agonists
|
Partly effective as agonists, no matter the amount employed
|
|
|
Inverse Agonists
|
Stabilize the receptor in its inactive conformation
(Clinically….. There really are no such Inverse Agonists) |
|
|
Receptor Occupancy Theory
|
- There is a certain # of receptors that need to be bound for a certain effect
- Clark attempted in the 1920s to quantify drug effects through application of the law of mass action. Out of his efforts and the contributions of others emerged the occupation theory of drug action. T- he occupation theory holds that the magnitude of a pharmacologic response elicited by a drug that reversibly combines with its receptor is directly proportional to the number (or fraction) of receptors occupied by the drug. |
|
|
EC50
|
concentration of drug that produces 50% of the maximal response
|
|
|
competitive antagonists
|
- Drugs that bind reversibly to a receptor at the same site as the agonist but have an intrinsic activity of zero (no receptor activation; a = 0) are competitive antagonists.
- By making receptors less available for agonist binding, a competitive antagonist depresses the response to a given dose or concentration of agonist. -The result is a parallel shift to the right of the agonist dose-response curve. - The most important aspect of this type of inhibition is that it is completely surmountable by a sufficiently high dose of agonist. - As in enzymology, the presence of a competitive antagonist produces an apparent reduction in the affinity of an agonist for its receptor. |
|
|
Non-competitive agonists
|
- The noncompetitive blockade is insurmountable in that the ceiling effect of an agonist can never be reattained, regardless of the dose administered.
- A noncompetitive antagonist may decrease the effective number of receptors by irreversibly binding to the receptor site or binding with such affinity that the agonist cannot successfully compete with it for binding. - The result is a downward displacement of the agonist log dose-response curve. |
|
|
Allosteric Antagonist
|
Drug produces effect by binding a site on the receptor distinct from that of the primary agonist and thereby changing the affinity of the receptor for the agonist
--> resembles non-competitive antagonism |
|
|
Action of drugs not mediated by receptors
|
- Agents that are chemically reactive (i.e. antacids)
- Agents that act according to non-pharmacological colligative properties (Cholesterol-binding agents - orally) - Drugs that are structural analogs of normal biological chemicals - incorporated into cellular components and thereby alter their function (analogs of pyrimidines and purines) |
|
|
In humans, the ______ represents the major pathway for drug excretion.
|
kidney
|
|
|
xenobiotics
|
Usually: lipophilic chemicals that in the absence of metabolism would not be efficiently eliminated
(environmental pollution, food additives, cosmetic products, agrochemicals, processed foods and drugs) |
|
|
Most drugs are metabolized in the
|
liver
|
|
|
The most common way to metabolize a drug is
|
during distribution
|
|
|
Metabolism during absorption....
|
One of the most important things that can occur during oral administration is the first-pass effect
--> BEFORE being distributed, the drug is metabolized This means we will have LESS of the drug reaching the blood… and the receptors (DECREASE IN BIOAVAILABILITY) --> Therefore, must take MORE of the drug to get same effect |
|
|
What methods don't really undergo 1st pass metabolism?
|
sublingual & rectal routes
|
|
|
For hepatic metabolism... most of the reactions are caused by _____ ...... and the reaction types are mostly ______
|
enzymes / oxidation
|
|
|
Hepatocyte
|
extensive network of smooth endoplasmic reticulum that catalyzes the metabolism of a variety of endogenous chemicals
|
|
|
Phase 1 of metabolism is responsible for
|
inactivation of the drug
(does LITTLE to affect the water solubility o the drug) --> cytochrome P450 is the most important enzyme in phase 1 |
|
|
Phase 2 of metabolism is responsible for
|
increase water solubility & mol. weight --> drugs get eliminated more easily
facilitate the elimination of drugs and the inactivation of the electrophilic and potentially toxic metabolites produced by oxidation |
|
|
What is the most important enzyme in phase 1
|
cytochrome P450 (catalyzes oxidation of the drug to be eliminated)
|
|
|
oxidation results in compounds that are:
|
- more polar
- relatively more hydrophilic - LESS LIKELY to penetrate cells and bind to tissue elements |
|
|
common things that affect cytochrome p450
|
- Grapefruit juice inhibits cytochrome P450 so unable to metabolize the drug
- char-grilled meat INDUCES so the drug works better and is better able to metabolize |
|
|
CYP34A
|
- CYP34A is responsible for 28% of the liver… but is responsible for the metabolism of ~ 60% of the drugs
|
|
|
reduction reactions
|
- limited to molecules with nitro or carbonyl groups or azo linkages.
- most reductions appear to result primarily from the action of enteric bacteria - when reduction occurs at one site in a molecule, oxidation usually takes place elsewhere, and the final product is more polar despite the initial addition on hydrogen atoms |
|
|
hydrolysis reactions
|
occasionally depends on microsomal enzymes
(not too important) |
|
|
Dehalogenation rxns
|
compounds such as: chlorophenothane and some volatile general anesthetics (halothane, sevoflurane) - dehalogenated by microsomal enzymes
may result in the formation of potentially toxic metabolites (not too important) |
|
|
Glucuronide conjugation
|
- only phase 2 reaction catalyzed by microsomal enzymes (glucuronosyltransferases)
- the conjugate is usually excreted by active secretion, into the bile or urine - is a quantitatively significant metabolic pathway for many drugs and their metabolites --> morphine: primary mode of metabolism (- Since phase 2, responsible for making drugs MORE hydro-soluble so it will NOT be reabsorbed) |
|
|
difference between 1st pass metabolism and enterohepatic cycle
|
1st pass metabolism in the beginning when you LOSE drug (happens during absorption)
Enterohepatic cycle at the end.. You actually GAIN drug (happens during metabolism) **enterohepatic cycle is the reason why you can only take birth control once a day |
|
|
factors that might interfere with the metabolism
|
- liver condition
- enzymatic inhibition/induction - genetic factors - age - pathology - cardiac insufficiency - diet and environmental factors |
|
|
Cardiac insufficiency
|
decrease the cardiac flow --> therefore decrease the liver clearance
If you have less blood, or the blood is moving slower ---> would take longer for the drug to reach the liver and be metabolized (which means you have too much active drug) |
|
|
Prodrugs
|
Drugs that become active after metabolism
may change: - Effect - Toxicity - Pharmacokinetics – absorption, distribution |
|
|
Clearance
|
body’s efficiency in eliminating drug
Amount of drug removed by the kidney per unit of time CL = rate of elimination / concentration of drug |
|
|
Volume of distribution
|
apparent space in the body available to contain the drug
**If drug has a high volume of distribution… this means that that the drug might be very liposoluble and would be found in the fat, NOT in the blood |
|
|
Elimination half-life
|
rate of removal of drug from the body
|
|
|
Bioavailability
|
fraction of drug absorbed as such into the systemic circulation
|
|
|
If drug has a high volume of distribution… this means that that the drug might be very _____and would be found in the ______
|
If drug has a high volume of distribution… this means that that the drug might be very liposoluble and would be found in the fat, NOT in the blood
|
|
|
Volume of distribution may vary widely depending on:
|
- The relative degrees of binding to high-affinity receptor sites
- Tissue proteins - The partition coefficient of the drug in fat - Accumulation in poorly perfused tissues |
|
|
If you have a drug that has a very high MW or bound to a plasma protein, most of the drug would be in the blood volume of distribution would be ____
|
LOW
|
|
|
One-compartment model
|
Body - single homogeneous compartment
- Does not describe the entire time course of the plasma concentration - Sufficient to apply to most clinical situations for most drugs (Its almost like the body is just the blood) |
|
|
Multi-compartment model
|
- For many drugs, the simple single-compartment model does not adequately describe the early time course of plasma concentration.
- Large discrepancies are particularly likely to be observed when a relatively lipophilic drug is given intravenously, as in the use of CNS depressants for conscious sedation. - In that situation, the assumption that the body acts as a single compartment does not hold, and one or more additional drug reservoirs must be postulated. --> our body is actually a multi-compartment system |
|
|
HALF-LIFE
|
Time it takes for the plasma concentration or the amount of drug in the body to be reduced by 50%
** Halflife is dependent on clearance If you have a pt. who has a disease that increases clearance ...half-life will be lower |
|
|
MEC
|
minimal effective concentration for the desired result
|
|
|
- Oral administration ____________ bioavailability of the drug
|
decreases
|
|
|
Steady State
|
- Attained after approximately four half-lives
- Time to steady state independent on the dosage(constant dose). - Steady state occurs when the amount of drug administered (in a given time period) is equal to the amount of drug eliminated in that same period. -At steady state the plasma concentrations of the drug (Css p) at any time during any dosing interval, as well as the peak and trough, are similar. |
|
|
Time to steady state is _____ of the dosage(constant dose).
|
independent
|
|
|
Loading dose
|
- You don’t increase the dose in every dose… JUST the beginning, and then you give maintenance dose
- When you give a loading dose reach steady state MUCH FASTER |
|
|
If the you give the same amount of drug to a patient who is deficient in plasma proteins in the drug, will :
|
have much more free drug in the blood b/c not enough plasma proteins to bind to
|
|
|
Hypoalbuminemia
|
Less drug binding to albumen (plasma protein) will increase amount of free drug !!!
(Reduce the extent of binding of acidic and neutral drugs) |
|
|
Phenytoin
|
- anticonvulsant drug
- more than 90% bound to plasma protein |
|
|
How would you adjust to Hypoalbuminemia?
|
1) Adjust the dose (Lower dose)
2) Shorter dosing intervals Therapeutic plasma levels |
|
|
Adverse Effects of Impaired Hepatic Clearance of Drugs
|
- decrease the metabolism of some drugs (Less blood going to the liver = less metabolism)
- increase the oral bioavailability of drugs - marked increase - drugs that normally have high first-pass hepatic clearance **First pass effect--> you will lose some of the drug b/c will be metabolized by the liver So if out of the original 100, the liver normally takes out 30, so you would have 70 in the blood… if the liver isnt functioning properly, and only takes out 20, you would have 80 remaining in the blood |
|
|
How does liver disfunction relate to first pass effect?
|
First pass effect --> you will lose some of the drug b/c will be metabolized by the liver
- So if out of the original 100, the liver normally takes out 30, so you would have 70 in the blood… if the liver isnt functioning properly, and only takes out 20, you would have 80 remaining in the blood Therefore, if drugs have high first-pass hepatic clearance, you probably need to modify / stop them |
|
|
How can you see how much the liver is metabolizing?
|
- There is NO quantitative way to see how much the liver is metabolizing
- So what you have to do… is titrate the dose.. Start small and increase and see if a pt responds (usually thru IV not oral) ideally measure plasma concentration in hospital |
|
|
Adverse Effects of impaired renal clearance of drugs
|
elevated levels of drug
- Renal system is responsible for most elimination of drug via urine - If not eliminated, still have drugs in body, some may be active some inactive… - One way to monitor this is amount of creatinine in the urine --> compare with normal value |
|
|
Drugs to be considered for pt. with Impaired HEPATIC Clearance of Drugs
|
- morphine
- meperidine - midazolam - nifedipine |
|
|
Drugs to be considered for pt. with Impaired RENAL Clearance of Drugs
|
- Vancomycin
- Aminoglycoside antibiotics - Digoxin |
|
|
What should be employed when renal clearance of a drug is diminished
|
- DECREASE the dose
- INCREASE the interval between doses |
|
|
how would you estimate glomerular filtration rate ?
|
- Iothalamate clearance method
- Creatinine clearance |
|
|
Optimal therapeutic decisions
|
Based on the evaluation of the individual patient in concert with assessment of the evidence for efficacy and safety of the treatment under consideration
|
|
|
Total body water for infants are __%, adults are__%
|
Total body water for infants are 80%, adults are 50-60%
|
|
|
Gastric pH of newborn
|
Gastric pH = 7-8 at birth and then falls to < 3 during the first 1-2 h of life
|
|
|
hepatic enzymes in children
|
Hepatic enzymes don’t work very well in children don’t even HAVE cytochrome P450
|
|
|
young kidney
|
- Young kidney is less competent to excrete drugs
|
|
|
Pharmacokinetic changes in the elderly population
|
Results from changes in body composition and the function of drug-eliminating organ.
|
|
|
changes in the elderly
|
- Clearance of many drugs - reduced
- Renal function - decline - 50% of that in young adults - Hepatic blood flow and drug metabolism - reduced - Elimination half-lives of drugs - increased (Elimination for half lives is increased because takes much longer for drug to leave the body) |
|
|
receptor sensitivity in the eldery
|
- Physiological changes and loss of homeostatic resilience - increased sensitivity to unwanted effects of drugs
- More illnesses - Consume a disproportionate share of prescription and over-the-counter drugs + age-related changes = serious adverse effects and drug interactions. |
|
|
malformation of the unborn child
|
1st trimester
|
|
|
functional disturbances in growth and maturation (when in development of unborn child?)
|
after 1st trimester
|
|
|
problems during birth
|
will affect the neonate (newly born child)
|
|
|
Parts of the Central Nervous System
|
- brain
- spinal cord |
|
|
Parts of the Peripheral Nervous System
|
- Somatic Nervous System
- Autonomic Nervous System - Sympathetic - Parasympathetic - Enteric |
|
|
main function of the autonomic nervous system
|
Maintenance and control of an stable internal environment (homeostasis), as an answer for fluctuations in the internal conditions and variations induced by external stimulations.
|
|
|
sympathetic activation of the heart
|
increase the heart rate, contractility and conduction velocity
|
|
|
sympathetic activation of the lung
|
increase respiratory frequency
|
|
|
sympathetic activation of blood vessels
|
skin and mucosa = constriction
Skeletal muscles = dilation |
|
|
sympathetic activation of the eyes
|
- Pupil dilation (mydriasis);
- relaxation of the ciliary muscle for far vision |
|
|
mydriasis
|
Pupil dilation
|
|
|
sympathetic activation of the adrenal medulla
|
secretion of Epinephrine and Norepinephrine
|
|
|
miosis
|
pupil constriction
|
|
|
parasympathetic activation of the eyes
|
- Pupil constriction (miosis)
- Lens accommodation to a short focal distance |
|
|
parasympathetic activation of blood vessels
|
- Vasoconstriction of the leg muscles
- Vasodilatation of the viscera (internal organs) |
|
|
Pre-ganglionic neuron
|
- cell body in brain or spinal cord
- axon is myelinated type B fiber that extends to autonomic ganglion |
|
|
Post-ganglionic neuron
|
- cell body lies outside the CNS in an autonomic ganglion
- axon is unmyelinated type C fiber that terminates in a visceral effector |
|
|
Sympathetic Fibers
|
- pre-ganglionic fiber is SHORT / post is LONG
- Some fibers go from the spinal cord to the adrenal medulla, activating it and causing a discharge of epinephrine into the blood |
|
|
Parasympathetic Fibers
|
- Preganglionic fiber is long
- Post ganglionic is shorter (because already close to target organs) |
|
|
where would you find acetylcholine ? (symp or para?)
|
BOTH
In Sympathetic… Ach is usually pre-ganglionic In Parasympathetic, ONLY have Acetylcholine |
|
|
where would you find norepinephrine? (symp or para?)
|
Norepinephrine ONLY in sympathetic
|
|
|
in stressful situations, what neurotransmitter is released?
|
norepinephrine (which is present ONLY in sympathetic response)
|
|
|
cholinergic vs. adrenergic neurons
|
Classified as either cholinergic or adrenergic neurons based upon the neurotransmitter released
Adrenegic --> adrenaline (NE) Choligernic --> Acetylcholine |
|
|
adrenergic receptors
|
a class of G protein-coupled receptors that are targets of the catecholamines, especially norepinephrine (noradrenaline) and epinephrine (adrenaline).
(G- proteins need 2nd messengers, so we need less drug molecules binding to those receptors to actually have many cell responses) |
|
|
The main types of adrenergic receptors are:
|
alpha and beta receptors
Alpha1 and Beta1 receptors produce excitation Alpha2 and Beta2 receptors cause inhibition Beta3 receptors (brown fat) increase thermogenesis |
|
|
compare the effects triggered by adrenergic neurons compared to those triggered by cholinergic neurons
|
Effects triggered by adrenergic neurons typically are longer lasting than those triggered by cholinergic neurons..
|
|
|
Alpha1 and Beta1 receptors produce _______
|
Alpha1 and Beta1 receptors produce excitation
Alpha2 and Beta2 receptors cause inhibition Beta3 receptors (brown fat) increase thermogenesis |
|
|
Alpha2 and Beta2 receptors cause __________
|
Alpha1 and Beta1 receptors produce excitation
Alpha2 and Beta2 receptors cause inhibition Beta3 receptors (brown fat) increase thermogenesis |
|
|
Beta3 receptors increase ___________
|
Alpha1 and Beta1 receptors produce excitation
Alpha2 and Beta2 receptors cause inhibition Beta3 receptors (brown fat) increase thermogenesis |
|
|
What is the MOST IMPORTANT way to stop the effect of NE that’s in the synaptic cleft
|
MOST IMPORTANT way to stop the effect of NE that’s in the synaptic cleft is through RE-UPTAKE
- NE will go back to the neuron, where it may be stored in vesicles again or destroyed by mono-amino oxidase (MAO), which is an intra-neuronal enzyme that destroys NE - NE in the synaptic cleft can also go thru uptake by the effector organ and go into cells or blood; can be destroyed by an enzyme called catechol-O-methyl transferase (COMT)…It is widely distributed in many tissues and is the principal extraneuronal enzyme involved with the metabolic inactivation of norepinephrine. |
|
|
MAO
|
During re-uptake, NE will go back to the neuron, where it may be stored in vesicles again or destroyed by mono-amino oxidase (MAO), which is an intra-neuronal enzyme that destroys NE
|
|
|
COMT
|
One method of reuptake is...NE in the synaptic cleft can also go thru uptake by the effector organ and go into cells or blood; can be destroyed by an enzyme called catechol-O-methyl transferase (COMT)…It is widely distributed in many tissues and is the principal extraneuronal enzyme involved with the metabolic inactivation of norepinephrine.
|
|
|
receptors for epinephrine
|
alpha-1
alpha2 beta-1 beta-2 Epi drug will cause BOTH constriction in the skin/mucosa AND relaxation in the skeletal muscles |
|
|
receptors for norepinephrine
|
alpha-1
alpha-2 beta-1 NE will ONLY cause constriction of the skin/mucosa (NO EFFECT ON RELAXATION OF SKELETAL MUSCLES) |
|
|
alpha-1 receptor
|
constricts the skin & mucosa
|
|
|
beta-2 receptor
|
dilates skeletal muscles
|
|
|
effects of epi vs. NE
|
NE does NOT bind to beta-2
- Epi drug will cause BOTH constriction in the skin/mucosa AND relaxation in the skeletal muscles - NE will ONLY cause constriction of the skin/mucosa (NO EFFECT ON RELAXATION OF SKELETAL MUSCLES) |
|

|

|
|
|
Effects of Epinephrine
|
Epinephrine affects alpha and beta receptors
- stimulate heart (B-1) - relax lung (B-2) - constrict skin & muscosa vessels (A-1 & A-2) - Relax skeletal muscles (B-2 & A-1) |
|
|
Effects of Norepinephrine
|
norepinephrine affects alpha and beta-1 reeptors
- stimulate heart (B-1) - NO EFFECT on lung (B-2) - constrict skin & muscosa vessels (A-1 & A-2) - CONSTRICT skeletal muscles (B-2 & A-1) |
|
|
Effects of Isopre
|
isopre affect beta receptors
- stimulate heart (B-1) - relax lung (B-2) - NO EFFECT on skin & muscosa vessels (A-1 & A-2) - Relax skeletal muscles (B-2 & A-1) |
|
|
Muscarinic receptors
|
- acetylcholine receptors that form G protein-receptor complexes in the cell membranes of certain neurons and other cells.
- They play several roles, including acting as the main end-receptor stimulated by acetylcholine released from postganglionic fibers in the parasympathetic nervous system. |
|
|
Cholinergic neurons release
|
Cholinergic neurons release acetylcholine
- all pre-ganglionic neurons - all parasympathetic post-ganglionic neurons - few sympathetic post-ganglionic neurons (to most sweat glands) (Excitation or inhibition depending upon receptor subtype and organ involved) |
|
|
compare uptake of andergenic vs. muscarinic/nicotinic receptors
|
adrenergic receptors have reuptake
nicotinic/muscarinic DO NOT (or very little) |
|
|
Acetylcholine crosses the junctional cleft and attaches _______to the postjunctional receptor, which exists in close proximity to a highly specific enzyme, ___________. Acetylcholine becomes bound to the enzyme at two primary sites and is hydrolyzed to _____ and ______ at such a rapid rate that the nerve can respond to another stimulus milliseconds later.
|
Acetylcholine crosses the junctional cleft and attaches reversibly to the postjunctional receptor, which exists in close proximity to a highly specific enzyme, acetylcholinesterase (AChE). Acetylcholine becomes bound to the enzyme at two primary sites and is hydrolyzed to choline and acetate at such a rapid rate that the nerve can respond to another stimulus milliseconds later.
(Acetylcholine is also removed from the junctional cleft by the simple process of diffusion.) |
|
|
Even in the total absence of AChE activity, the action of acetylcholine can be terminated quickly by pseudocholinesterase, a nonspecific plasma enzyme also known as ___________, which is found in many tissues, including blood.
|
butyrocholinesterase
|
|
|
butyrocholinesterase
|
Even in the total absence of AChE activity, the action of acetylcholine can be terminated quickly by pseudocholinesterase, a nonspecific plasma enzyme also known as butyrocholinesterase, which is found in many tissues, including blood.
|
|
muscarinic receptors
|

|
|
|
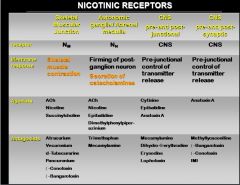
nicotinic receptors
|
- Form ligand-gated ion channels
- Do not operate with a second messenger - Forms a pentameric structure consisting of homomeric a and b subunits. - In humans, eight a and three b subunits have been cloned. |
|
|
Nicotinic receptors usually cause contraction of the ________ and secretion of ___________.
|
Nicotinic receptors usually cause contraction of the skeletal muscles and secretion of catecholamines (Epi or NE) in the adrenal medulla
|
|

|
|
|
|
Sympathomimetics
|
Sympathomimetics = mimics the sympathetic response
|
|
|
Sympatholytics
|
Sympatholytics = antagonists; will have an opposite effect of the sympathetic response
|
|
|
Parasympathomimetics
|
Parasympathomimetics = mimic the parasympathetic response
|
|
|
Parasympatholytics
|
Parasympatholytics = antagonists; have opposite effects of the parasympathetic response
|
|
|
Dominance by the sympathetic system is caused by ____________
|
Dominance by the sympathetic system is caused by physical or emotional stress --
“E situations” - emergency - embarrassment - excitement - exercise |
|
|
flight or fight response
|
- dilation of pupils
- increase of heart rate, force of contraction & BP - decrease in blood flow to nonessential organs - increase in blood flow to skeletal & cardiac muscle - airways dilate & respiratory rate increases - blood glucose level increase |
|
|
why are sympathetic responses long-lasting?
|
Long lasting due to lingering of NE in synaptic gap and release of norepinephrine by the adrenal gland
|
|
|
rest-and-digest
|
parasympathetic responses
- Mechanisms that help conserve and restore body energy during times of rest - Normally dominate over sympathetic impulses |
|
|
SLUDD type responses & 3 decreases
|
associated with parasympatheric responses
salivation, lacrimation, urination, digestion & defecation 3 “decreases” = 1) decreased HR 2) diameter of airways 3) diameter of pupil |
|
|
muscarinic receptors --> nerves
|
receptor = M1, M2
response = DECREASE ganglionic transmission |
|
|
muscarinic receptors --> Heart
|
receptor: M2, M3
Response: vasodilation < cardiac rate < rate of conduction in the specialized tissues of the SA and AV nodes < force of cardiac contraction |
|
|
muscarinic receptors --> Smooth Muscles
|
Receptors: M2, M3
Response: > GIT tone & motility > amplitude of contraction > secretory activity Detrusor - contraction > voiding pressure > ureter peristalsis |
|
|
muscarinic receptors --> Glands
|
Receptors: M1, M3
Responses: INCREASE secretions |
|
|
muscarinic receptors --> CNS
|
Receptors: M1, M2, M3. M4, M5
Response: > cognitive function > seizure activity < dopamine release M4 and M5 - facilitate dopamine release |
|
|
nicotonic receptors --> skeletal muscular junction
|
Receptor: Nm
Membrane Response: skeletal muscle contraction Agonists: Ach, Nicotine, Succinychloline |
|
|
nicotonic receptors --> autonomic ganglia/ adrenal medulla
|
Receptor: Nn
Membrane Response: firming of the post-ganglion neuron - secretion of catecholamines Agonists: ACh, nicotine, epibatidine |
|
|
Adrenergic Agonists
|
Drugs that mimic or enhance the actions of norepinephrine or epinephrine in adrenergic neurotransmission
Effect - usually similar to the sympathetic nervous system stimulation |
|
|
What are the possible fates of norepinephrine by ending the synapse?
|
1) negative feedback given to the neuron, which tells body to stop the release of NE into the synaptic cleft
2) re-uptake to the neuron.. where it will be destroyed by MAO ... or restored in vesicles 3) taken up by effector organ and COMT breaks down NE |
|
|
Sympathomimetics are much related to ______
|
Sympathomimetics are much related to BP, heart and lungs (main uses for the drug)
|
|
|
main actions of aympathomimetics
|
- Stimulates a / b receptors
- Increases the NE release with or without direct stimulation |
|
|
alpha 1 drugs
|
- Eyes – mydriasis, or if blocked, can cause miosis
- BV – skin and mucosal BV. Drug binding causes constriction - Bladder – changes the muscle and sphincters |
|
|
alpha 2 drugs
|
- Causes relaxation/dilation of vessels
- Found in presynaptic terminals to inhibit NE pre-neuronally - Found in effector organs, then causes dilation of vessels |
|
|
what are the main receptors for skin and mucosa?
|
alpha-1
|
|
|
beta 1 drugs
|
Primarily in the heart (stimulates)
|
|
|
Beta-2
|
- Mostly in the lung and cause broncodialtion / relaxation
- lDilation of skeletal muscle vessels |
|
|
classification of adrenergic agonists
|
- receptor affinity
- receptor localization - mechanism of action |
|
|
classifying mechanism of action of adrenergic agonists
|
Direct-acting --> directly binds to receptor
Indirect-acting --> no binding but somehow increases the endogenous E or NE Mix-acting --> does both, binds to receptor and releases NE |
|
|
chart of classifying adrenergic agonists
|
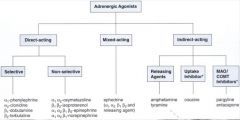
|
|
|
direct-acting adrenergic agonists
|
Drugs acting directly on the adrenergic receptors (agonists)
response: similar to the sympathetic stimulation (SAME EFFECT AS THE ENDOGENOUS COMPOUND IN THE BODY) |
|
|
We use a direct-acting adrenergic agonist as an anesthetic in dentistry... what do we want from that?
|
We want to decrease the absorption of lidocaine (You want to work with the longer period of time)
|
|
|
What are the IMPORTANT direct-action adrenergic agonist drugs?
|
P.E.N.I.L.
Propranolol --> beta-1 & beta-2 Epinephrine --> alpha-1, alpha-2, beta-1, beta-2 Norepinephrine --> alpha-1, alpha-2, beta-1 Isoprenaline --> beta-1 & beta-2 Levonordefrin --> alpha-1 alpha-2, beta-1 |
|
|
vascular effects of norepinephrine
|
Local injection: vasoconstriction
Intravascular injection: Constriction of ALL vessels Increases - peripheral resistance Increases BP - in physiological doses Baroreflex - slows the heart. |
|
|
vascular effects of a local injection of epinephrine
|
vasoconstriction
|
|
|
vascular effects of a local injection of norepinephrine
|
vasoconstriction
|
|
|
Intravascular injection of norepinephrine
|
Constriction of ALL vessels
|
|
|
Intravascular injection of epinephrine
|
DEPENDS ON THE DOSE !!!
Low dose of EP - Decrease BP - more B-2 receptors active → decrease in BP High dose EP - similar to NE effect: - Increase in the BP |
|
|
vascular effects of epinephrine
|
Local injection: vasoconstriction
Intravascular injection: Epi - mixed effects - DOSE Decreases - skin, mucosa, and kidneys blood flow rate In therapeutic doses - it doesn’t affect the brain blood flow rate Increases - Lung blood pressure Increases - coronary blood flow rate Skeletal muscles - vasodilation slightly decreases - Blood Pressure (BP) and tachycardia |
|
|
high vs. low doses of epinephrine
|
high dose --> ENHANCE the blood pressure
- Direct stimulation - myocardium - Increase the cardiac frequency - Vasoconstriction low dose --> DECREASE the blood pressure - Stimulation of beta 2 receptors from the skeletal muscle vessels - dilation |
|
|
- In dentistry, what do we use with local anesthetic?
|
a vasoconstrictor
--> this will help to keep the anesthetic localized - Increase duration – slower absorption - Decrease toxicity - slower absorption, and will be metabolized and eliminated - Decrease bleeding – vasoconstrictor thus causes hemostasis |
|
|
stress will increase endogenous levels of
|
epinephrine and noepinephrine
|
|
|
Systolic blood pressure results from ________
|
the cardiac stimulation by beta 1
(the heart) |
|
|
Diastolic blood pressure results from _____
|
the vessels resistance by alpha 1 and beta 2
(blood vessels) |
|
|
pharmacological effect of epi & NE on the heart
|
- Direct excitation - beta 1 receptors in the myocardium, and of cells of the pacemaker and conducting tissues.
- Cardiac systole is shorter and more powerful, cardiac output is enhanced, and the work of the heart and its oxygen consumption are markedly increased. - Cardiac efficiency is lessened. |
|
|
baroreceptors
|
Once you have a change in the BP or heart rate --> baroreflex --> Slows down the heart (after E / NE acts)
|
|
|
blood pressure in response to giving a bolus injection of EPI or NE:
|
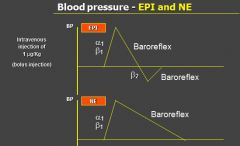
- High dose administration of EPI → BP increases because it is binding to a1 and b1 causing the constriction of the vessels
- the resulting high blood pressure is decreased via the baroreflex |
|
|
blood pressure in response to epi in low doses
|
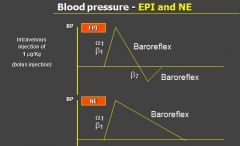
- B2 action occurs and there is a slight decrease in BP
- Baroreflex recognizes this and INCREASES HR |
|
|
blood pressure in response to norepinephrine
|
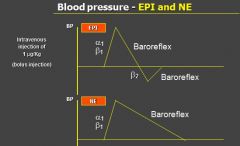
- no action on B2 so there is no decrease in BP, thus we just see an INCREASE in BP and then baroreflex will bring it back down (red circled portion in Epi)
|
|
|
vasoconstrictor in the blood can cause:
|
- Slight decrease in BP if we administer the drug slowly
- Slow administration = low dose = no absorption ---> Safe for a hypertensive or cardiac diseased pt even if accidently administered into the blood |
|
|
effects of epi on GIT and bladder
|
- Relaxation - smooth muscles
- Reduction - Tone and the frequency and amplitude of spontaneous contractions - Contraction - sphincters - In vitro contraction of the uterus / Close to the due day - relaxation - Relaxation - bladder detrusor and contraction - bladder sphincter --> retention of materials inside |
|
|
effects of epi on metabolism
|
- Increase glucose level via glycogenolysis
- Inhibit insulin secretion → more glucose available - Cells in the brain do not require insulin (I assume she means GLUTs and etc. to transport glucose via insulin receptors) --> Can just directly transfer glucose to the inside of the cells |
|
|
Isoproterenol
|
adrenergenic agonist --> beta agonist
- Increases heart rate, force of contraction, and cardiac output with no reflex vagal activation (direct beta 1 effects) - Vasodilation in skeletal muscle, resulting in decreased diastolic BP and decreased TPR (beta 2 effects) Uses: - Torsades de points (heart disease) - Cardiac arrest of complete heart block - Asthma or COPD |
|
|
Dobutamine
|
adrenergenic agonist --> beta-1 agonist
- Increase contractility and heart rate Uses: - Severe congestive heart failure **this is good b/c no changes in the lungs or anywhere else except for in the heart |
|
|
Albuterol
|
andrenergic agonist --> beta-2 agonist
Uses: - Bronchodilator - long-term treatment of obstructive airway diseases, asthma, and for the treatment of acute bronchospasm |
|
|
Penylephrine
|
andrenergic agonist --> alpha- agonist
- Marked arterial vasoconstriction during intravenous infusion. - May activate b-receptors - at much higher concentrations. - Neo-Synephrine Uses: - restore blood pressure during spinal or general anesthesia, in hypotensive emergencies, or after overdose of an antihypertensive medication - nasal decongestant |
|
|
Clonidine
|
andrenergic agonist --> alpha-2 agonist
- Capacity to lower blood pressure - activation of alpha 2 receptors in the cardiovascular control centers of the CNS - Changes in blood pressure and heart rate - post-synaptic a2 receptors in many blood vessels - Transient vasoconstriction - followed by a more prolonged hypotensive response = decreased sympathetic outflow from the CNS Uses: - Hypertension, antidepressive |
|
|
what is important about clonidine
|
Therapeutic effects, but dangerous as A2 is found in the preneuronal membrane
- A2 agonist, hence, the effect is inhibition of more release of Epi Do NOT bind to other organs receptors → inhibits release of NE (although NE levels not high) - Anti-adrenergic |
|
|
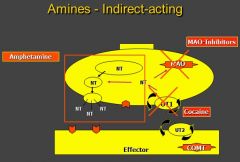
what are indirect acting adrenergic agonists
|
Drugs that do not bind to the receptor but increase NE in the synapse
|
|
|
indirect acting adrenergic agonists
|
- Stimulation of the mediator synthesis and release (amphetamine)
- Inhibition of the mediator uptake (cocaine) - Inhibition of the mediator inactivation (MAO Inhibitors) ***Non - selective thus have lots of side effects but we have been using these for so long that in some patients, body finds homeostasis with these drugs |
|
|
amphetamine
|
an indirect acting adrenergic agonist
Stimulation of the mediator synthesis and release of norepinephrine Uses: - Attention deficit hyperactivity disorder (amphetamine) - Drug abuse Tolerance: - Readily develops to the CNS stimulant effects, appetite suppression and mood elevation |
|
|
cocaine
|
an indirect acting adrenergic agonist
--> stimulates release of NE - Prevents reuptake and no therapeutic effect whatsoever - Inhibition of the mediator uptake and thereby prolong the effect of the adrenalines |
|
|
MAO inhibitor
|
indirect acting adrenergic agonist
--> increases amount of NE - Inhibition of the mediator inactivation - MAO = Enzyme to breakdown NE, prolonged effect achieved by inhibiting MAO Anti-depressant drugs |
|
|
what is the most important example of a mixed-acting adrenergic agonist?
|
ephedrine
|
|
|
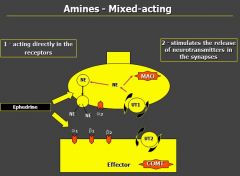
mixed-acting adrenergic agonists
|
Two different mechanisms
- Directly in the receptor (a and b agonist) - Releases mediators in the synapses Ex. Ephedrine |
|
|
- Amphetamine and MAO inhibitors Increases the release of NE
- Drugs inhibit reuptake of EPI such as cocaine - No selectivity - Work in the CNS and ANS as well as every single organ - Therefore, it does have some therapeutic uses, but the patients need to be monitored closely due to this non-selectivity |
|
|
- ephedrine
- increased release of NE in synapse |
|
|
Adverse Reactions of Adrenergic Drugs
|
- Dose related: too large dose, accidental intravascular injection, impaired uptake of the drug, heightened sensitivity or number of adrenergic receptors, preexisting cardiovascular disease
- Patients with history of uncontrolled hyperthyroidism, hypertension, or angina pectoris --> susceptible to cardiac disturbances - local tissue necrosis |
|
|
Why is it important for us as dentists to give vasoconstrictors with local anesthetics?
|
By adding vasoconstrictors, you can almost double the duration of the LA, and do NOT need to administer a higher dose
--> helps to prevent LA toxicity |