![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
184 Cards in this Set
- Front
- Back
|
CD marker for Hematopoeitic Stem Cells |
CD 34+ |
|
|
Myeloid & Lymphoid Lineages |
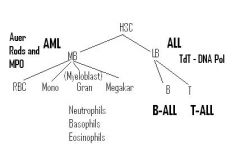
Myeloblast & Lymphoblast Lineages |
|
|
Normal WBC and the Names for High & Low WBC |
5-10k Leukocytosis >10k & Leukopenia <5k |
|
|
What cells can contribute to high/low WBC count? |
Any WBC can contribute to high or low WBC count (Only WBC's not RBC's or platelets)
however,
Only 1 type of WBC is usually contributing to the change in WBC |
|
|
Neutrophilic Leukocytosis |
High Number of Neutrophils causing WBC >10k |
|
|
Alkylating agents side effects |
Chemotherapy agents (Destroy proliferating (rapidly-dividing) cancer cells by adding an alkyl group to Guanine-> Preventing DNA replication)
-> Neutropenia (b/c Neutrophils also rapidly dividing)
also a risk factor for Acute Myeloid Leukemia arising from Myelodysplastic Syndrome (b/c division/differentiation/downstreamvprolifertion is impaired, mutated Myeloblasts w/ acquiredmutations proliferate -> >20% of BM) |
|
|
Basophilia |
CML (almost only time seen) -> b/c Basophils represent smallest % of WBC's in blood, even few during infection. The only exception is CML!!! |
|
|
Monocytosis seen in: |
1) Chronic Inflammatory States - B/c MPh modulate Chronic Inflammation
2) Malignancy -
|
|
|
Gram negative sepsis can cause what in neutrophil count? |
Decreased Neutrophil Count due to mobilization to tissues |
|
|
Why do Corticosteroids Cause Neutrophilic Leukocytosis |
They do not Increase total # of Neutrophils in body, but they cause demargination of Neutrophils attached to endothelial cells in Pulmonary Blood Vessels |
|
|
Causes of Neutrophilic Leukocytosis |
1) Bacterial Infection & 2) Tissue Necrosis Both 1 &2 trigger Acute Inflammation-> stimulated BM to rapidly release Granulocytes -> So rapidly that some are immature ->Left Shift-> Decreased Fc Receptors (nencessary for opsinization)
3) High Cortisol State - cause Neutrophils marginated to pulmonary vasculature to demarginate and enter circulation |
|
|
High cortisol state |
Causes apoptosis of lymphocytes->Lymphopenia |
|
|
What cell line is most sensitive to whole body radiation? |
Lymphocytes
|
|
|
Fc Receptors?
|
CD16 |
|
|
Effect of Severe infection on Neutrophil Count |
Neutropenia b/c severe infection draws most Neutrophils out of blood & to the site of infection |
|
|
What distinguishes immature neutrophils? |
Low expression of Fc receptors(CD16) |
|
|
Lymphopenia caused by: |
1) Immunodeficiency ->Underproduction of Lyphocytes (e.g. DiGeorge Syndrome ->missing thymus->Tcells don't develop properly)
2) High Cortisol State (Induces Apoptosis of Lymphocytes)->Cushing's Syndrome (High amount of Cortisol)
3) Autoimmune destruction (e.g. SLE destryos many blood cell types)
4) Whole Body Radiation - Lymphocytes most sensitive cell to Radiation a) Chernobyl workers had severe Lymphopenia b) Knock out Auto-reactive lymphocytes w/ Radiation prior to BM Transplant |
|
|
How does bacterial infection cause leukocytosis |
1) Mobilization of marginal pool
2) BM stimulated to rapidly release Granulocytes
|
|
|
Myeloid Cell descendants |
|
|
|
Monocytosis is seen in |
chronic inflammatory state & malignancy |
|
|
Eosinophilia is seen in |
1) Allergic hypersensitivity
2) parasitic infection
3) Hodgkin's Lymphoma |
|
|
Mechanism of Eosinophilia in Hodgkin's lymphoma... |
increased IL-5 production by neoplastic cells |
|
|
Basophilia |
Chronic Myelogenous Leukemia(CML) exclusively - b/c physiologically Basophil count is very low |
|
|
Eosinophilia |
1) Allergic Reactions
2) Parisitic Infections
3) Hodgkin's Lymphoma (some pts Have Eosinophilia due to Tumor producing IL-5) |
|
|
Lymphocytic Leukocytosis is seen in... |
↑ circulating lymphocytes due to: 1) Viral infection - b/c viruses are obligate/intracellular so they are killed by CD8+ Tcells
2) Bordetella pertussis (exception b/c bacteria)- produces Lymphocytosis Promoting Factor that blocks Lymphocytes from getting to/stimulated to fight infection in Lymph nodes ->they end up being stuck in blood->Lymphocytosis |
|
|
Lymphocytic leukocytosis is a/w what BACTERIAL infection? MOA |
Bordetella Pertussis |
|
|
Infectious Mononucleosis |
Characterized by Reactive CD8+ Tcells
historically mistaken for Monocytes (Therefore the name Mononucleosis is a Historic MISTAKE)
|
|
|
Types of ALL |
B-ALL (B-lymphocyte precursor)
T-ALL (T-lymphocyte precursor)
Both will be TdT+, but will have different surface markers |
|
|
Causes of Infection Mononucleosis |
1) EBV 2) CMV - less common cause |
|
|
How does EBV spread & what tissues does it infect? |
Teenagers spread via saliva
Infects:
1)Oropharynx epithelial cells -> sore throat
2) Hepatocytes- hepatitis and hepatomegaly, elevated liver enzymes
3) B cells |
|
|
Bodies Response to EBV |
1) Generalized LAD (Enlarged Lymph Nodes) - B/c viral proteins presented on MHC Class I of oropharyngeal epithelium/hepatocytes/Bcells, resulting in proliferation of CD8+ Tcells in lymph nodes->Hyperplasia of Paracortex of Lymph Nodes (Paracortex = where Tcells reside, Cortex = B-cell Territory)
2) Splenomegaly - Hyperplasia of PALS (Peri-Arterial Lymphatic Shealth) in White Pulp (Red pulp - RBC's, White Pulp - Blue Lymphocytes)-> White Pulp expanded b/c Tcells surround arterial blood vessels in White pulp-Peri-Arterial Lymphatic Sheath (very blue)
3) High White Count w/ (due to) Atypical Lymphocytes - Reactive CD8+ Tcells that look Atypical b/c they look like Monocytes
|
|
|
ALL Most Common in: |
All Children < 5y.o. or Down's children who are >5y.o. |
|
|
atypical lymphocytes (which look like monocytes). They are reactive CD8+ T-cells that look like monocytes is characteristic of? |
viral infection |
|
|
Monospot test. Interpretation? |
Screening test - Detects IgM heterophile (Hetero-other animal RBC's) Ab's that agglutinate RBCs from another animal = Pathologists accidentally discovered that IgM Ab's generated in response to EBV happen to be reactive against Ag's on Sheep/Horse RBC's
Usually Positive 1 wk after EBV infection - b/c takes 1 wk to generate some Adaptive Immun.
|
|
|
EBV Complications |
1) Incr Risk of Splenic Rupture
2) Rash if Exposed to Pennicillin - Penicillin likely hapten
3) Dormancy of EBV virus in Bcells leading to: a) Increased risk of recurrence 2) Incr risk of lymphoma esp. if pt is immunocomprimised - (e.g. AIDS) - probably b/c EBV causes changes in Bcell DNA & there are too few Tcells to identify this via MHC Class I and Induce Apoptosis of those Bcells |
|
|
Splenic rupture? |
Infectious mononucleosis with EBV b/c of Hyperplasia in PALS (Peri-arteriolar lymphatic sheath) |
|
|
Rash after penicillin |
EBV |
|
|
EBV risks |
increase the risk of B-cell lymphoma, especially if immunocompromised |
|
|
Leukemia |
Neoplastic Proliferation of Blasts They stop differentiating to downstream cells, but rather start accumulating |
|
|
How does Acute Leukemia Present? |
Acute Anemia, Thrombocytopenia, & Neutropenia
w/ HIGH WBC due to Blasts entering Blood
(Blasts->Large/Immature cells w/ Massive Nucleus/Lots of cytoplasm. Nucleolus looks like a punched out region in massive nucleus) |
|
|
How do we diagnose AML vs. ALL? |
Lymphoblasts will be TdT+ (DNA Polymerase necessary for genetic rearrangement in B/Tcells precursors) in Nucleus->ALL
Myeloblasts will have MPO (Myeloperoxidase)->AML
[MPO] is Highest in Azurophilic Granules ->Lysosomes of Neutrophils->Causes green/blue color in pus->produces Hypochlorous Acid (HOCl=bleach) from H2O2 & Cl- during Neutrophil's respiratory burst
|
|
|
Acute Leukemia |
>20% Blasts in BM->Crowd out Normal Hematopoesis!!!: If low RBC's ->Anemia->Hypoxia Low PMN's :Neutropenia->Infection Low Platelets: Thrombocytopenia-> bleeding
Nmally Blasts = 1-2% ->enough for Nml Hematopoesis |
|
|
Types of Leukemia |
ALL (Lymphoblasts) & AML (Myeloblasts) |
|
|
Characteristic marker for ALL? |
TdT+ = DNA polymerase exclusive to LymphoBlastic precursors
(Myeloblasts & Mature B/Tcells will be TdT-) |
|
|
Characteristic marker for AML? Exceptions? |
MPO and sometimes MPO crystallizing into Auer Rods (crystallized MPO) are exclusive to AML b/c they will not ever express MPO as they are not involved in Oxidative Burst Killing |
|
|
Children over 5 with Down's syndrome a/w |
ALL - b/c have extra Ch 21
Transolcation involving Chr 21 associated w/ ALL |
|
|
Most common type of ALL? Classic markers? |
B-ALL |
|
|
What prophylaxis do you need to administer in a case of B-ALL? |
B-ALL has EXCELLENT response to chemotherapy
CSF and scrotum require direct injection (prophylaxis) since they have a BBB & Blood-Testis Barrier that prevents chemo from getting to them b/c ALL will be in these tissues as well |
|
|
T-ALL markers?
|
CD2 through CD8 (in addition to TdT+) |
|
|
B-ALL chromosomal abnormalities?
|
t(12;21) translocation - good prognosis |
|
|
Common presentation of T-ALL |
T- Acute Lymphoblastic Lymphoma (NOT Leukemia) mediastinal Thymic mass in a Teenager |
|
|
Classic AML |
Acute Promyelocytic Leukemia -> t(15;17) (Promyelocytes = precursors of Granulocytes->Neutrophils, Eosinophis, Basophils)
Involves translocation/disruption of Retinoic Acid Receptor (RAR) on Chm. 17 to Chm 15;
RAR disruption/dysfunction due to change in structure: prevents differentiation/maturation of Promyeloblasts ->promyelocytes(blasts) accumulate-> These accumulating blasts contain lots of Auer Rods which can activated coagulation -> Risk of DIC->Medical Emergency |
|
|
APL puts you at risk for? How do you prevent this? |
DIC due to Auer rods in blasts. |
|
|
Acute Myeloid Leukemia arises predominantly in what demographic? |
Older population, 50-60 yrs ->b/c due to accumulated mutations/translocations rather than mistakes in rearrangement seen in lymphocytes (early in life during B/Tcell maturation) |
|
|
leukemia with infiltration of gums. Notable lab feature? |
Acute Monocytic Leukemia(AML subclassification)->Infiltrates Gums (swelling of gums) b/c Monocytes infiltrate tissues |
|
|
leukemia that lack MPO |
Acute Megakaryoblastic Leukemia (don't need/use MPO) |
|
|
Children under 5 with Down's syndrome a/w
|
Acute Megakaryoblastic Leukemia |
|
|
Hematopoietic Stem Cell expresses what receptor
|
CD34+ |
|
|
Chronic Leukemia is characteristic of what cell line? |
Neoplastic proliferation of naïve lymphocytes (B and T cells) |
|
|
Chronic Lymphocytic Leukemia has what cells? What receptors are expressed? |
B-cells. CD5 and CD20 |
|
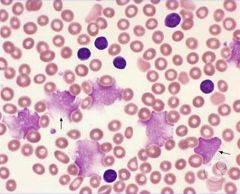
|
Smudge Cells classically seen in CLL |
|
|
Small Lymphocytic Lymphoma |
If in CLL the neoplastically proliferating Naive B/T-cells enter the lymph nodes forming a MASS
involvement of lymph nodes described as lymphodenopathy (lymph nodes are abnormal in size, number or consistency = swollen lymph nodes)
Once the naive B/Tcells form MASS in lymph nodes, that MASS is called:
Small Lymphocytic Lymphoma (small in size) this is in addition to existing CLL (Leukemia) |
|
|
Complications of CLL |
1) Hypogammaglobulinemia: Neoplastic B-cells can not make normal Ig's. As they take over, they represent the majority of B-cells, therefore norml B-cell number is low, and these CLL lymphocytes interfere with generating humoral immunity ->the few pts. that die usually die from Infection
2) Autoimmune Hemolytic Anemia - If these CLL B-CELLS actually produce Ab's they will be self-reactive->destroy self-RBCs
|
|
|
What is a risk factor for large B-cell lymphoma
|
CLL (Richter Transformation) |
|
|
Most common cause of death in CLL
|
infection due to defective Ig production |
|
|
tartrate resistant acid phosphatase |
TRAP...a/w Hairy Cell Leukemia |
|
|
hairy cytoplasmic processes a/w what disease? |
Tartrate resistant acid phosphatase (Hairy Cell leukemia - neoplastic proliferation of mature B-cells) |
|
|
Tx of hairy cell leukemia? |
2-CDA (cladribine) = Adenosine Deaminase Inhibitor
Adenosine Deaminase is part of purine degredation pathway |
|
|
ATLL |
Adult T-cell Leukemia\Lymphoma
|
|
|
geographic associations with ATLL |
Japan and the Caribbean |
|
|
Clinical features of ATLL |
1) Rash - B/c Tcell Leukemia cells often go to skin (just like normal Tcells = think Delayed Hypersensitivity)
2) Generalized Lymphadenopathy with Hepatosplenomegaly (B/c mature lymphocytes programmed to go to lymph node/spleen, but b/c there are so many in this neoplasia, they cause enlargement)
3) Lytic (punched out) bone lesions with hypercalcemia |
|
|
Mycosis Fungoides - clinical symptoms? |
Also Neoplastic proliferation of mature CD4+ T-cells (just like ATLL) = called Pautrier microabscesses (b/c Tcells go to skin)
Pautrier microabscesses = Mycosis Fungoides (name is a historic misnomer b/c thought it was infection)
|
|
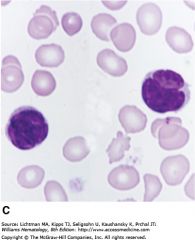
|
Sezary Cell -> Sezary Syndrome = when Neoplastic Mature Tcells of Mycosis Fungoides spread to BLOOD
Sezary cells = have Cerebriform nuclei (look like lobes of brain), mycosis Fungoides, CD4+ leukemia with skin infiltration (Pautrier Abscesses) |
|
|
Myeloproliferative Disorders |
Neoplastic proliferation of Mature cells of Myeloid lineage seen in late adulthood -All Myeloid cells will be expanded, but diseases named based on predominant cell that has proliferated
High WBC count (b/c always some granulocyte expansion = granulocytes are WBC's) w/ Hypercellular BM (b/c neoplastic proliferation)
|
|
|
MOA of myeloproliferative symptom of hyperuricemia and gout |
Increased turnover of cells result in activation of purine degradation pathway (e.g. in polycythemia vera, a lot of nuclei are removed from RBC's as they mature and the Nucleic acid has to be degraded) whose end metabolite is uric acid->uric acid crystals deposit in big toe ->cause gout
|
|
|
Chronic Myeloid Leukemia. Characteristic chromosome abnormality? What cell type is characteristically increase? |
Type of Myelodysplastic Disorder Neoplastic proliferation of Granulocytes
Notable increase in basophils t(9;22) - Ph Chrm -- generates BCR-ABL combination that increases tyrosine kinase activity |
|
|
First line treatment for CML? MOA? |
Imatinib which blocks the tyrosine kinase activity
Tyrosine Kinase (Growth Singnal Transducer = oncogene) |
|
|
Worst complication of CML? Preceded by? |
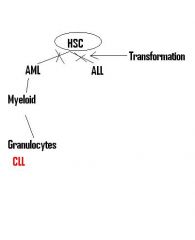
Acute Leukemia heralded by new onset enlargement of spleen due to mutation accumulation
If this happens, likely to continue accumulating ->transformation can turn into AML OR ALL since the mutation is in the HSC (CD34+) |
|
|
Distinguish between a leukemoid reaction(infection) & CML |
Since in Leukemoid Rxn, you can also expect proliferation of Granulocytes w/ Left Shift
CML will have the following findings: |
|
|
JAK2 Kinase Mutation |
1) Polycythemia Vera
2) Essential Thrombocythemia
3) Myelofibrosis |
|
|
Classic a/w Polycythemia Vera
|
most common cause of Budd-Chiari syndrome-- blockage of hepatic vein |
|
|
itching after bathing
|
Polycythemia Vera due to excessive mast cells |
|
|
treatment for polycythemia vera |
W/out Tx., DEATH in 1 year
1.) Phlebotomy - Decrease RBC's
2.) Hydroxyurea - effective agent for myelosuppression |
|
|
Reactive Polycythemia vs Polycythemia Vera |
Reactive Polycythemia is due to BM REACTING to high EPO levels
High EPO levels can be caused by
1) Lung disease-> Decreased SaO2, increased EPO
In Polycythemia Vera, the RBC production is not a reaction to EPO Increase, but rather a BM Neoplastic Proliferation |
|
|
Causes of Excessive Platelets? |
1) Essential Thrombocythemia
2) Iron deficiency anemia - probably b/c BM stimulated to make Myeloid cells in hopes of making RBC's, but can't make enough RBC's b/c can't make enough Hb |
|
|
Difference between Essential Thrombocythemia & other myelodysplastic syndromes? |
Unlike other Myelodysplastic Syndromes, Essential Thrombocythemia:
1) Rarely progresses to Marrow Fibrosis (b/c further down lineage so doesn't stimulate fibroblasts since Megakaryocytes & NOT platelets produce PDGF)
2) Rarely progresses to Acute Leukemia (b/c mutations are further down in lineage)
2) No risk for hyperuricemia/ gout (due to the fact there is not an increase in turnover of nuclear material since platelets are just pieces of megakaryocyte cytoplasm pinched off) |
|
|
JAK2kinase mutation associated with marrow fibrosis |
myelofibrosis |
|
|
Clinical features of myelofibrosis
What cell predominates? |
1) JAK2 Kinase mutation leading to proliferation of mature myeloid cells (particularly Megakaryocytes)
2) Neoplastic Proliferation of Megakaryocytes -> Producess Excessive PDGF->Marrow Fibrosis (therefore name: Myelo (Myeloid lineage) Fibrosis (BM Fibrosis)) [Normally, BM just has Fat & BM elements (cells)]
2) Splenomegaly due to extramedullary hematopoiesis (b/c BM Fibrosed), as HSC's move to spleen
3) LeukoErythroBlastic Smear - Normally: BM has Reticulin gates that prevent immature (BIG) cells from exiting into blood. When Hematopoesis shifts to Spleen b/c of BM Fibrosis, Spleen has no way to prevent Immature cells from entering blood-> Immature Leukocytes/Erythrocytes (Nucleated RBC's =Howell Jowelly bodies) & Blasts enter blood (seen on smear)
4) Increased Risk of Infection/Thrombosis/Bleeding b/c Spleen much smaller than total BM was, so can not keep up w/ Normal Production
5) Tear-drop RBC's ->The little Hematopoesis left in fibrosed BM, results in Tear-drop RBC's as these RBC's squeeze through Fibrosed Matrix |
|
|
tear drop RBC's a/w |
myelofibrosis |
|
|
follicular lymphadenopathy
|
Reactive Hyperplasia due to Rheumatoid arthritis and early HIV (due to HIV infection of dendritic cells expressing CD4+ receptors = found in follicles) |
|
|
Paracortex lymphadenopathy |
Reactive Hyperplasia due to viral infections |
|
|
Deep in medulla lymphadenopathy |
Reactive Hyperplasia due to Lymph Node draining tissue with cancer and reacting to Cancer Ag (doesn't imply metastasis) APCs (Histiocytes) within the sinus that filter the lymph, engulf neoplastic prticles and present them to lymphocytes. Lymphocytic rxn to these Ag, stimulates these Sinusoidal Histiocytes to proliferate
|
|
|
Small B cell lymphoma's (arise from neoplastic proliferation fo small B-cells) |
Follicular lymphoma Defined by their location in/around the follicle of Bcells in the Lymph node Cortex
|
|
|
Intermediate sized b cells lymphoma |
Burkitt lymphoma |
|
|
Most common form of non-hodgkin's lymphoma |
Diffuse Large B cell lymphoma |
|
|
What receptor do small b-cells express
|
CD20+ |
|
|
Chromosomal abnormality with follicular lymphoma |
Driven by t(14;18) = IgH, BCL2 translocation
BCL2 overstabilization of mitochondria causes failure of unreactive B-cells (those whose hypermutation lead to worse binding to Ag) to die (via apoptosis) during somatic hypermutation
B/c neoplastic Bcells in the follicle will not be able to be induced into apoptosis, they will proliferate->lymphoma |
|
|
Treatment for follicular lymphoma |
only for symptomatic people - Rituximab (anti-CD20 Ab - kills Bcells) + low dose chemotherapy |
|
|
demographic of small b cell lymphomas |
mostly late adulthood
as mutations or translocations accumulate |
|
|
Important complication of follicular lymphoma What else can progress to this? |
Progression to diffuse Large b-cell lymphoma
|
|
|
Follicular Hyperplasia vs Follicular Lymphoma |
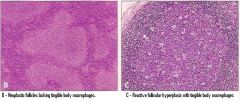
Important to distringuish b/c infection will Normally cause Hyperplasia of follicles (as B cells undergo Hypermutation) 1) Disruption of architecture (follicles stay near cortex in normal tissue but are everywhere (invading the whole Lymph node) in Lymphoma) |
|
|
Mantle Cell lymphoma chromosomal abnormality
gene product?
Pathology? Age of pts? |
t(11;14) which switches cyclin D1 gene with IgH chain. Cyclin D regulates progression from G1 -> S
Expansion of Bcells immediately around follicles
Like all other mature Bcell Lymphoma's ->older adults get it |
|
|
Marginal Zone Lymphoma |
Associated with chronic inflammatory states b/c if Margin arises (it only exists in cases of chronic Inflammation) it is the Memory Bcells that have gone through Hypermutation in the Follicle
In chronic Dz. they are repeatedly stimulated/divide until they accumulate Genetic Defect
such as
Hashimoto Thyroiditis,
Sjogren Syndrom ->after a long time 1 parotid gland elarges b/c Marginal Cell Lymphoma Bcells infiltrate 1 parotid and proliferate there)
H. Pylori gastritis - MALToma is a marginal zone lymphoma in mucosal sites. Could go away if you Tx H. Pylori infection
|
|
|
"Neoplastic proliferation of cells immediately adjacent to follicle"
|
Mantle Cell Lymphoma |
|
|
WBC's |
Leukocytes - All blood cells w/ the exception of RBC's & Platelets |
|
|
H. pylori gastritis can lead to |
Gastric MALToma, which is a marginal zone lymphoma if ulcer is chronic. If you tx. H. pylori the Gastric MALToma will often resolve |
|
|
Burkitt Lymphoma presents in what demographic?
|
Presents in child/young adult as an extra nodal mass. |
|
|
Burkitt Lymphoma geographical associations?
|
African form a/w jaw |
|
|
Burkitt's lymphoma chromosomal abnormality?
|
t(8;14) results in translocation of c-myc to the IgH leading to overexpression of c-myc. |
|
|
Burkitt's Lymphoma is a/w what infection?
|
EBV |
|
|
most common form of non-hodgkins lymphoma? |
Diffuse large b-cell lymphoma |
|
|
How can diffuse large b-cell lymphoma arise?
Who gets it and how does it present? |
Arises:
1) Sporadically OR 2) Transformation of Follicular lymphoma (as it accumulates more mutations)
Late adulthood as an enlarging lymph node or Extranodal Mass |
|
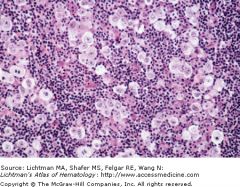
|
lacunar cells |
|
|
owls eye nuclei
|
reed-sternberg cell |
|
|
Mixed cellularity hodgkin's lymphoma is a/w?
|
eosinophilia and abundant eosinophils in tissue due to the production of IL-5 by reed-steinberg cells |
|
|
Neoplastic proliferation of plasma cells in Bones? What etiology? |
Multiple Myeloma - Most common primary malignancy of bone (Most common overall is metastatic carcinoma)
|
|
|
Multiple Myeloma clinical features?
|
1) production of osteoclast activating factor (IL-1B) causes lytic bone lesions(Remember ATLL) especially in vertebrae and skull
Light chains in urine:into the urine Bence-Jones protein and causes renal failure in the kidneys. |
|
|
What types of Ig is produced by multiple myeloma?
|
IgG and IgA |
|
|
Increased serum protein decreases charge between RBCs causing them to stick together...
|
Rouleaux formation of RBCs due to multiple myeloma |
|
|
Most common cause of death in multiple myeloma?
|
increased risk for infection due to overproduction of useless Ig(lacks antigenic diversity) |
|
|
What is diagnostic of Monoclonal Gammopathy of Undetermined Significance (MGUS)? |
Gammopathy = Plasma Cell Dyscrasia (Defect)
Only a finding of M-protein spike in electrophoresis but lacking any other feature of multiple myeloma... |
|
|
Waldenstrom's Macroglobulinemia |
B-cell lymphoma that produces monoclonal IgM(MACROglobulinemia b/c IgM is a Pentamer = it's a BIG Ig)
2) Increased Serum Proteins: M-spike due to IgM (Not IgG or IgA)
3) No lytic bone lesions (B/c Osteoclast Activating Factor= IL1B NOT Produced)
4) Oveload of neoplastic monoclonal expansion of Big IgM causes serum hyperviscosity ->
a) Visual & Neurologic Deficits due to retinal hemorrhage or stroke b) Bleeding - b/c hyperviscosity leads to defective platelet aggregation
leading to defective platelet aggregation |
|
|
What receptors on Hodgkin's lymphoma?
|
It is a B-cell lymphoma but NOTABLY does NOT have CD20 receptor. It has CD15 and CD30 and contains Reed-Sternberg cells which are associated with "owl's eye" |
|
|
classic presentation of nodular sclerosis hodgkins? |
young adult female with cervical or mediastinal lymph node |
|
|
What are langherhan's cells? |
1) Specialized dendritic cells found predominantly in skin
2) Derived from BM monocytes
3) Present Ag to NAive Tcells (they are APC's) |
|
|
Langerhans Cell Histiocytosis |
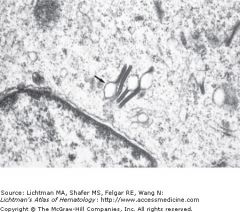
Neoplastic Proliferation of Langershans Cells
Birbeck granules (Tennis racket) seen on electron microscopy in these cells |
|
|
Chronic leukocytic leukemias |
B-cell Leukemia's |
|
|
Myeloproliferative Disorders
|
Chronic Myeloid Leukemia(CML) |
|
|
Lymphadenopathy |
Can be chronic(painLESS) or acute(painFUL) |
|
|
Name all the important Lymphomas |
Non-hodgkins |
|
|
Small Lymphoma(s)
|
--Follicular Lymphoma |
|
|
Large Lymphoma(s)
|
--Diffuse Large B cell lymphoma |
|
|
Intermediate Lymphoma(s)
|
-- Burkitt's Lymphoma
|
|
|
someone with CLL that has an acutely enlarging lymph node/spleen
|
Richter transformation -- progression of CLL to Diffuse large B-cell lymphoma |
|
|
skin rash with lytic bone lesions
|
A-TLL |
|
|
lytic bone lesions
|
multiple myeloma |
|
|
"starry sky"
|
burkitt's lymphoma |
|
|
most common PRIMARY malignancy of bone?
|
multiple myeloma - malignant proliferation of plasma cells in the bone marrow |
|
|
Most common subtype of Hodgkin's lymphoma? |
Nodular Sclerosis |
|
|
These diseases will have M spikes on serum protein elecrophoresis...
|
Multiple Myeloma |
|
|
How is Prognosis for B-ALL generated? |
Based on Cytogenetic Abnormality (based on outcomes of past pts. w/ a given abnormality) |
|
|
Types of B-ALL Cytogenetic Abnormalities & their outcomes? |
t(12,21) 1) Most common 2) Seen in kids 3) GOOD PROGNOSIS
t(9,22) ->Ph+ ALL 1) rare for B-ALL ->t(9,22) is defining for CML 2) Seen in adults 3) POOR Prognosis 4) t(9,22) known As Philadelphia Ch.->Ph+ALL |
|
|
Average Age of AML pts. |
Older: 50-60 y.o. |
|
|
How to Subclassify AML |
Most preferred in descending order Based on:
1) Cytogenetic Abnormalities - predominant way
2) Lineage of Myeloblasts
3) Surface marker |
|
|
Tx for Acute Promyelocytic Leukemia |
ATRA- All Trans Retinoic Acid -> can bind/stimulate defective RAR that resulted from t(15,17) allowing Promyelocytes to differentiate to Granulocyte Descendents (Eosinophils, Basophils, Neutrophils)->resolves leukemia/Danger of DIC due to Auer Rod initiated coagulation |
|
|
How do you classify AML in absence of recurrent cytogenetic Abnormality? |
Based on Lineage of Differentiation->e.g. Erythroblastic AML, Myelobastic AML, Megakaryoblastic AML... |
|
|
Leukemia Risk in Down's Syndrom based on age |
<5 y.o. Acute Megakaryoblastic Leukemia is a proliferation of Megakaryoblasts (type of AML) = we do not know why...
>5 y.o. ALL (translocation of 12:21 more likely w/ extra Ch 21) |
|
|
Why Lymphoblastic Leukemia seen in young & Myeloblastic Leukemia seen in old |
B/c Lymphocytic rearrangement done to generate huge Adaptive Immunity done early in life->so likely translocations to occur early on
Myeloblastic Leukemia involves non-rearranging Innate Immunity, so mutations/transolcations more likely accumulate w/age |
|
|
AML can arise from what pre-existing Dysplasia? |
Myelodysplastic Syndrome =
Arises from mutations in Myeloblast
Differentiation of blood precursor cells is impaired
significant increase in levels of apoptotic cell death in bone marrow cells
Clonal expansion of the abnormal cells results in loss of ability to differentiate
When overall percentage of bone marrow myeloblasts rises over cutoff (20%), then by definition = (AML)
Progression of MDS to AML is example of the multi-step theory of carcinogenesis = series of mutations occur in an initially normal cell and transform it into a cancer cell |
|
|
What causes Myelodysplastic Syndrome |
Alkylating Agents (Chemo) & Radiotherapy Disrupt normal Hematopoesis->Cytopenias and accumulation fo Disrupted (Dysplastic) Blasts in BM
B/c the % of these Dysplastic Blasts is <20% it is not classified as a Acute Leukemia, but once it passes to >20% it is called AML |
|
|
What do most Myelodysplastic Syndrome pts. die from? |
Infection or bleeding due to cytopenias->
Bleeding from Thrombocytopenia Infection from Neutropenia |
|
|
What is possible progression if a lot of mutations accumulate in Myelodysplastic Syndrome Blasts? |
Acute Leukemia (Blasts>20%) |
|
|
Why was chronic Leukemia given its name? |
B/c the neoplastic proliferation of mature lymphocytes in the blood that is insiduous (as opposed to acute) in onset (b/c involves the slow accumulation of mutations as opposed to rapid changes due to translocations in blasts)
and the consequences are also slowly developing as opposed to acute cytopenias ans sometimes even asymptomatic and can LIVE VERY LONG TIME W/ DISEASE, b/c no major risk of infection/hypoxia seen in ALL
|
|
|
clinical features of Hairy Cell Leukemia |
1) Splenomegaly - notably, expansion of RED PULP (where RBC's normally are) as opposed to expected White pulp
3) Lymphadenopathy is ABSENT because they are trapped in red pulp.
Hairy Cell Leukemia-> think TRAP - "Bcell pulling its Hair B/c it's TRAPPED"
1) TRAP + = (titrate Resistant Acid Phosphatase) 2) Cells get TRAPped in Red Pulp -> Splenomegaly 3) TRAPped in BM where Hairy Cells synthesize/Assemble fibronectin matrix= BM fibrosed ->Dry tap 4) B/c TRAPped in Spleen/BM, can't go to lymph node = No Lymphodenopathy |
|
|
Complications of Myeloproliferative Disorders |
|
|
|
Why can CML progress to AML OR ALL? |
B/c the translocation 9:22 = Ph Ch occurs in HSC's that have begun differentiation to Myeloid Lineage, and the Myeloblast then produces a lot of Myeloid cells
HOWEVER, if one of the Rapidly dividing neoplastic HSC w/ limited Myeloid differentiation accumulates more mutations, it can then dedifferentiate enough to result in proliferation of of either Myeloblasts (AML) or Lymphoblasts (ALL) and be arrested in that state |
|
|
3 Phases of CML |
1) Chronic - Stable Splenomegally
Can progress to...
2) Accelerated - Enlarging Splenomegally suggests that enough mutations acquired to Accelerate proliferation and usually followed by->
3) Transformation phase = Develop Acute Leukemia (AML or ALL) (worst possible complication of CML) |
|
|
Likelihood of CML progressing to:
AML? ALL? |
AML (2/3) = 67%
ALL (1/3) = 33%
|
|
|
Polycythemia Vera |
Neoplastic proliferation of myeloid cells (including granulocytes/platelets) = myeloproliferative disorder BUT RBC's proliferate most
JAK2 kinase (a tyrosine kinase) mutation DRIVES Polycythemia Vera |
|
|
Clinical Symptoms of Polycythemia Vera |
Symproms caused by Hyper-Viscosity caused by thickening of blood due to RBC overload:
1) Blurry Vision & Headache 2) Increased risk of Venous Thrombosis -> Budd-Chiari Syndrome (thrombosis of Hepatic v. causes infarction of liver) 3) Flushed face (b/c hyperviscous blood congests in blood vessels of face) 4) Itching after Bathing b/c marked increase in Mast Cells, in addition to RBC's. The mast cells release histamine when bathing-> itching |
|
|
Essential Thrombocythemia |
Neoplastic proliferation of mature myeloid cells (including RBC's/Granulocytes) BUT ESPECIALLY: Platelets
Associated w/ JAK2 Kinase mutation (like Polycythemia Vera) |
|
|
Clinical Symptoms of Essential Thrombocythemia |
1) Bleeding (there might be many platelets but they are neoplastic so can be DEFECTIVE) or Thrombosis (if proliferating platelets are working)
|
|
|
Painless Lymphadenopathy |
When Lymph node drains site of Acute Infection (b/c acute Inflammation has PGE2 = sensitizes pain neurons) |
|
|
Painful Lymphadenopathy |
When Lymph node has expansion that does not involve Acute Inflammation
1) Chronic Inflammation (Autoimmunde Dz.)
2) Metastatic Carcinoma
3) Lymphoma |
|
|
Regions of Lymph Node Follicle |
Follicle is an accumulation fo B-cells that waits for Ag's as lymph drains from various tissues around the body
In case of infections, the follicular B-cells will become activated and form a Germinal Center where the reactive B-cells will undergo hypermutation (those poorly reactive to Ag will undergo apoptosis), and transform to Plasma Cells that will generate Ig
Surrounding the Germinal Cells is the Mantle Zone: small unchallenged B cells surrounding pale staining germinal centers
Marginal Zone: Typically only forms in cases of Chronic inflammation as it contains post-Germinal Center (have already gone through initial stimulation) memory B cells |
|
|
Why do Chronic INflammatory states lead to Marginal Zone Lymphoma |
B/c this is a neoplastic proliferation of Memory B-cells. SInce Chronic inflammation involves repeated stimulation/division of Memory B-cells, these cells are more likely to have a translocation of IgH chain and Cyclin....resulting in unabated proliferation of these cells |
|
|
Which major type of Lymphoma type is more common? |
Non-Hodgkin's Lymphoma = 60%
Hodgkin's Lymphoma = 40% |
|
|
Ways of Classifying Non-Hodgkin's Lymphoma's |
1) Cell Type B vs T cell
2) Cell Size - small, Intermediate, or LARGE
3) Pattern of Cell growth - do they grow as Follicles or Diffuse sheet?
4) Expression of Surface Markers (Bcell vs Tcell)
5) Cytogenetic Translocations |
|
|
Are vast majority of Non-Hodgkin Lymphoma's Bcell or Tcell Lymphomas? |
B-cell Lymphomas |
|
|
Are Small or Large Lymphocytes more differentiated? |
Small ones, Large ones are more Blast-like |
|
|
Burkitt Lymphoma |
Intermidiate Size neoplastic Bcells (more dedifferentiated than small Bcell lymphomas)
EXTRANODAL MASS in CHILD or Young Adult
Driven by translocation of c-myc (Nuclear regulator that activates many genes that promote growth) from Ch8 to Ch14 (IgH)->drives aberrant proliferation->production of tumor
2 types
1) African - Bcell expansion -> Jaw mass = associated w/ malaria & EBV (malaria increases suceptibility to EBV
2) Sporadic Form = usually abdominal mass |
|
|
Burkitt's Lymphoma Pathology |
High mitotic rate due to c-myc overexpression->Starry-Sky Appearance
SKY - Sheet of Neoplastic Bcell overgrowth
Stars - Non-staining areas where Tingible Body Macrophages are clearing up rapidly dividing Bcells that necrosed due to lack of O2 and nutrients
|
|
|
Diffuse Large B-cell Lymphoma |
Diffuse = Grows diffusely (Does NOT produce follicles, margin, mantle)
Large = Poorly Differentiated -> Clinically Very Aggressive
Most common Non-Hodgkins Lymphoma
|
|
|
What makes Hodgkin's Lymphoma DIFFERENT from Non-Hodgkin's Lymphoma |
It is NOT a MASS of Neoplastic cells
BUT
a few Ree-Sternberg Cells that secrete Cytokines attracting reactive WBC's = Mass of a few Reed-Sternberg cells surrounded by reactive (non-neoplastic) WBC's
|
|
|
How do Reed-Sternberg Cells look? |
Large Bcell w/ Multilobed Nucleus & Prominent Nucleoli
Owl-Eyed Nucleus = 2 Nuclei -eyes, each w/ dark Nucleolus-pupil. Owl= b/c close together
CD15+ & CD30+ BUT CD20- (despite being Bcells) |
|
|
Possible Consequence of Cytokines released by Reed-Sternberg Cells |
1) Occasionally B-Symptoms: Fever/Chills/Night Sweats
2) Attract reactive Lymphocytes, Plasma Cells, Macrophages, & Eosinophils (Make up Bulk of Mass)
3) May Lead to Fibrosis (Nodular Sclerosis) |
|
|
Subtypes of Hodgkins Lymphoma, & how are they classified? |
Based on prevelance of given Reactive cells
1) Nodular Sclerosis (70% of Hodgkin's Lymphoma Cases)
2) Lymphocyte Rich - tons of Reactive Lymphocytes attracted
3) Mixed Celularity - Mix of many WBC's
4) Lymphocyte Depleted - Few Lymphocytes amongst reactive WBC's |
|
|
Nodular Sclerosis |
Most Common Type of Hodgkin's Lymphoma
Classically - 1) Enlarged Neck Lymph node 2) Enlarged Mediastinal Lymph node
The pt. is often : A young adult Female |
|
|
Nodular Sclerosis Pathology |
Lymph node divided by Broad bands of Fibrosis that are dividing lymph node into Nodules
THerefore:
Nodular = Look like Nodules surrounded by Fibrosis
Sclerosis = Hardening b/c fibrotic tissue
Reed-Sternberg cells sit in big open spaces = called lakes->called Lacunar Cells = Nodular Sclerosis |
|
|
Which Hodgkin's Lymphoma Subtype has Best Prognosis |
Lymphocyte Rich |
|
|
Which Hodgkin's Lymphoma Subtype has WORST Prognosis |
Lymphocyte Depleted (Seen in ELderly and HIV+ Individuals = both Immunocompromised) |
|
|
Which Hodgkin's Lymphoma associated w/ abundant Eosinophils? |
Mixed Cellularity due to Reed-Sternberg Cells secreting lots of IL-5. These pts. can also have Eosinophilia from Reed-Sternberg Cells over-secreting l IL-5 |
|
|
What determines clinical problems in Multiple Myeloma? Give examples. |
Clinical Prolems Determined by what the Neoplastic Plasma Cells are Producing
1) Osteoclast Activating Factor (IL-1B) activates RANK Receptor on Osteoclasts = causes lytic "punched out lesions" in Verterbra & Skull (Big enough to be seen on X-ray) ->Bone Pain, Hypercalcemia (b/c bone Broken down), Increased Risk of Fracture (b/c bone weakened)
2) Ig ->High Serum protein (M spike = usually monoclonal IgG or sometimes IgM, seen on Serum Protein ElectroPhoresis (SPEP)
Increased Risk of Infection b/c Monoclonal Plasma cells are all making same Ab ->this monoclonal Ab ha no Ag Diversity->Infection->Most COmmon cause of DEATH
Rouleaux Formation - RBC's stacked like coins, b/c High Serum Protein due to monoclonal IgG, causes loss of charge on RBC's->stacking instead of interacting w/ H2O in blood that would make them dissolve well
3) Often Overproduction of Light chain relative to Heavy CHain->free Light chain enters serum, deposits in tissues, changes confirmation to Beta-Pleaded sheets (AL Amyloid= called primary Amyloidosis)
Proteinuria - If light chain secreted in urine (the light chains in urine are called = Bence-Jones Proteins)
Renal Failure - if Light chains deposit in Renal Tubules and cause damage = called myeloma kidney |
|
|
How do you Tx. Waldenstrom's Gammaglobulinemia? |
Plasmapharesis: Remove IgM from the Serum |