![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
147 Cards in this Set
- Front
- Back
|
Iron absorption occurs in the? |
duodenum |
|
|
Iron consumed in two dietary forms |
Heme form(meat) and non-heme form(vegetables) |
|
|
Describe transport and storage of iron |
1) Enterocytes take up Fe from gut lumen
2) Presence of ferroportin (transporter) on the basolateral side is the regulatory step that determines if Fe is absorbed into blood
3) Iron is then bound to transferrin in bloodstream where it is delivered to hepatocytes and bone marrow macrophages (MUST be bound to prevent ROS generation via Fenton rxn: Fe2+ + H2O2 ----> Fe3+ + ~OH + OH-)
4) Stored intracellular iron is bound to ferritin (also prevents ROS formation) |
|
|
Causes of Microcytic Anemia |
Problem w/ Nomral Hb production 1) Iron deficiency Anemia - Fe deficiency
2) Anemia of Chronic Disease - Fe locked up in Macrophages
3) Sideroblastic Anemia - protoporphyrin def.
4) Thalassemia - Globin defect |
|
|
Most common nutritional deficiency in the world? |
Iron deficiency |
|
|
Most important regulatory step in iron uptake? |
Ferroportin - transport of iron into blood (located on basolateral surface of enterocyte) |
|
|
TIBC |
measures amount of transferrin in blood |
|
|
Serum Iron |
Measure of iron in blood (this iron WILL be bound to transferrin) |
|
|
% Saturation |
percentage of transferrin bound to Fe |
|
|
Serum Ferritin |
amount of iron bound in liver and BM macrophages |
|
|
Lab findings in 1st stage of iron deficiency |
Storage iron is depleted 1st
↑ TIBC b/c liver makes more transferrin to try and pick up more Fe |
|
|
Lab findings in 2nd stage of iron deficiency |
Serum iron is depleted next (2nd)
% Sat of transferrin ↓ |
|
|
Lab findings in 3rd stage of Fe deficiency |
Normocytic anemia |
|
|
Lab findings in 4th stage of iron deficiency |
Microcytic, hypochromic anemia
Erythroblasts divide extra time (Microcytic) in hopes of maintaining close to Normla [Hb] |
|
|
clinical features of iron deficiency |
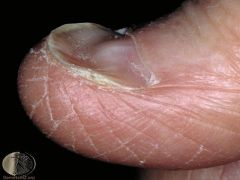
1) Anemia 2) Koilonychia (spoon shaped nails) 3) Pica (chew on abnormal items->eat dirt or other things in search of Fe) |
|
|
Describe RDW & FEP in Iron deficiency anemia... |
↑ RDW (RBC Distribution Width) ->spectrum of size of RBC's) due to initial normocytic RBC's mixing with microcytic, so the distribution of width is larger than Normal |
|
|
Plummer-Vinson syndrome |
1) Fe deficiency Anemia presents w/
2) Esophageal webs (obstruction in esophagus due to protrusions of mucosa-> poorly chewed foods can get stuck->dysphagia) &
3) Atrophic glossitis (smooth tongue) |
|
|
Patient presents with anemia, dysphagia, beefy red tongue |
Plummer-Vinson syndrome |
|
|
Anemia of Chronic Disease pathophys |
chronic disease --> chronic inflammation --> ↑ acute phase reactant: Hepcidin |
|
|
Iron deficiency occurs in cases of? |
1) Nutritional Deficiency
2) Blood loss |
|
|
Causes of Nutritional Deficiency of Fe |
1. Dietary insufficiency
2. Malabsorption
3. Gastrectomy - ↓Acid b/c part of stomach removed -> less Fe2+ (bioavailable b/c absorbable) since Acid necesary for reducing Fe3+ to Fe2+ |
|
|
Stages of Fe Deficiency |
1) Storage Fe depleted (b/c erythroblasts use up stores for Hb production) -> ↓ferritin BUT ↑TIBC (b/c liver senses low Fe stores and produces more transferrin to pick up any available Fe)
2) Serum Fe ↓ and % Saturation of Ferritin ↓ (18% instead of Nm: 33%) b/c serum Fe used
3) Normocytic Anemia - VERY EARLY STAGE OF Fe deficiency Anemia = Erythroblast can't make enough Hb, so just makes fewer cells, but they are normal in size/[Hb]
4) Microcytic/Hypochromic Anemia - As Fe defeciency becomes severe, much less Hb is made, so Erythroblasts divide extra times (forming smaller RBC's) in order to preserve minimum [Hb]. However, it will still be less than Nm -> Increased zone of central pallor due to ↓[Hb] |
|
|
Tx for Fe deficiency Anemia |
1) Find out cause (e.g. if Elderly Male: rule out carcinoma of colon)
2) Supplement Fe (Ferrous sulfate) |
|
|
Populations suceptible to Fe Deficiency |
1) Infants - Breast feeding (milk is low in Fe)
2) Children - poor diet
3) Adults - ♀- menorrhagia (blood loss) & pregnancy (deficiency b/c fetus uses a lot of Fe) ♂- peptic ulcer dz. (blood loss)
4) Elderly - Rich countries: colon polyps/carcinoma Developing countries: hookworm (Necatur/Ancylostoma) due to blood loss |
|
|
Most common cause Anemia in Hospitalized Pts. |
Anemia of Chronic Disease |
|
|
Anemia of Chronic Dz. caused by: |
Chronic Inflammation - Can be chronic infection but often Autoimmune
Cancer - will also trigger Chronic Inflammation |
|
|
Why body evolved to release Hepcidin during Chronic inflammation |
B/c body lacks mechanism to differentiate Chronic Inflammation induced by Infection vs. Autoimmune/Cancer->
sequesters Fe that could be used by bacteria to grow/divide->
to limit spread of infection-> Consequence: Microcytic Anemia
Happens in chronic inflammatory states even if no actual infection exists |
|
|
Lab findings in anemia of chronic disease |
↑ ferritin (b/c all Fe is bound up to ferritin)
↓ TIBC - b/c Liver ↓Transferrin levels when Ferritin levels are HIGH ↓ serum Fe - b/c Erythroblasts use up a lot of Fe from blood
↓ %sat - b/c a lot of Fe removed from Transferrin
↑ FEP (Free Erythrocyte Protoporphyrin b/c ↓ heme due to low available Fe) |
|
|
Tx for Anemia of Chronic Dz |
1) ↓ Inflammation-> ↓Hepcidin-> Erythroblasts can use stored Fe
2) EPO administration b/c Hepciding caused ↓EPO. This is particularly helpful for cancer pts. (probably b/c additional EPO downregulation mechanisms exist) |
|
|
Sideroblastic Anemia pathophys |
defective protoporphyrin synthesis
Fe will be transferred from BM Macrophages to Erythroblast Mitochondria and get stuck there b/c Protoporphyrin Deficiency interferes w/ Heme production |
|
|
Rate limiting step in protoporphyrin synthesis? |
In Erythroblast Cytosol
Conversion of: Succinyl-CoA -> Aminolevulinic Acid (ALA)
by the enzyme: Aminolevulinic Acid Synthetase (ALAS)
|
|
|
Second Major Step of Heme Synthesis |
In Erythroblast Cytosol
Conversion of: Aminolevulinic Acid(ALA) -> Porphobilinogen
by the enzyme: ALAD (ALA Dehydrogenase) |
|
|
Final step of protoporphyrin synthesis? Where does it occur? |
In Erythroblast Mitochondria
Attachment of: Protoporphyrin + Fe -> Heme
by the enzyme: Ferrochelatase |
|
|
Classic histological finding in sideroblastic anemia? What causes them? |
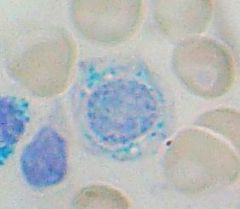
Ringed Sideroblasts
-Fe trapped b/c deficiency of functional Protoporphyrin |
|
|
What stain marks iron? |
Prussian Blue |
|
|
Congenital form of sideroblastic anemia caused by? |
Enzyme defect in ALAS (Aminolevulinic Acid Synthetase): the rate limiting step in Protoporphyrin/Heme Synthesis |
|
|
Acquired form of sideroblastic anemia caused by? |
1) Alcohol - Mitochondrial poison (dissolves Mitoch. membranes) that damages production of Protoporphyrin
2) Lead poisoning - Denatures ALAD & Ferrochelatase
3) Vitamin B6 deficiency - ALAS cofactor |
|
|
What drug commonly causes vitamin B6 deficiency? |
Isoniazid treatment (1. INH binds/inactivates pyridoxine 2. INH inhibits the enzyme that activates pyridoxine) |
|
|
Lab findings in sideroblastic anemia... |
Fe Overloaded state in Erythroblast Mitoch. Massive Fe stores generate ROS ROS damages erythroblast->cell lysis Fe leaks out Fe picked up by BM MΦ's-> Results in ↑ Ferritin-> ↓ TIBC B/c some Fe leaks out into blood stream-> ↑ serum Fe and ↑% sat
|
|
|
Another Fe Overloaded State w/ very similar lab findings to Sideroblastic Anemia |
Hemochromatosis (so much Fe that it is out of proportion to Normal levels of Protoporphyrin) |
|
|
What disease are people who are carriers for thalassemia protected against? |
Plasmodium falciparum malaria |
|
|
3 types of hemoglobin
|
Alpha 2, Beta 2 - adult (HbA) |
|
|
genetics of alpha thalassemia... |
4 total alpha globin alleles - 2 on each copy of chromosome 16 |
|
|
alpha thalassemia types common on what continents? |
cis-deletion of alpha alleles-Asia (DELETIONs on same Ch 16 copy (e.g. both on maternal Ch 16))->more likely to lead to Alpha Thalassemia Major->explains Higher incidence of miscarriage in Asia |
|
|
HbH |
Beta subunit tetramer (B/c insufficient copies of Alpha globin)
can be seen on electrophoresis |
|
|
HbBarts
|
Gamma subunit tetramer (B/c insufficient copies of Alpha globin) |
|
|
genetics of beta thalassemia |
problem due to gene MUTATION of 1 of 2 alleles of Beta Globin (1 on each copy of Chromosome containing them) |
|
|
isolated increase in HbA2
|
Beta Thalassemia Minor |
|
|
Hepatosplenomegaly with evidence of hematopoiesis in facial bones and skull |
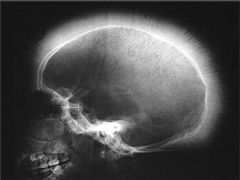
Massive Erythroid Hyperplasia |
|
|
Aplastic crisis |
due to parvovirus B19 infection in Beta Thalassemia Major patients
parvovirus B19 invades and destroys RBC precursors and halts the red cell production. Since Beta Thallassemia Major already causes few functional RBC's, this additional destruction causes Aplastic Crisis (severe anemia) |
|
|
Target cells
|
present in Beta Thalassemia Minor & Major
also post splenectomy b/c cells w/ extra cytoplasm are not removed/destroyed by Splenic MΦ's |
|
|
Target cells in thalassemia a result of...
|
Low levels of Hb in the RBC. This is like "taking the air out of a basketball" and allows the center to bleb out. |
|
|
Characteristic of Beta Thalassemia Major |
Nucleated RBC's - b/c RBC's are pushed out before completely mature b/c of Severe anemia |
|
|
Electrophoresis findings for Beta Thalassemia Major |
no HbA |
|
|
Macrocytic Anemia |
due to folate or vitamin B12( cobalamin) deficiency (megaloblastic anemia) |
|
|
hypersegmented neutrophils |
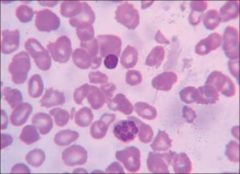
neutrophils with greater than 5 lobes |
|
|
Epithelial cell change in Folate/B12 deficiency |
Megaloblastic change in all rapidly dividing cells |
|
|
A drug that can cause macrocytic anemia
Clinical diseases that can cause it? |
5-Fluorauracil - Thymidylate synthase (TS) inhibitor
Interrupting the action of TS blocks synthesis of the pyrimidine thymidine, which is required for DNA replication
Alcohol and Liver disease can also cause macrocytic anemias but for completely different reasons
Alcohol - Directly damages BM
in Liver disease - Lecithin—cholesterol acyltransferase (LCAT) activity may be decreased (LCAT converts free cholesterol into cholesteryl ester (a more hydrophobic form of cholesterol), which is then sequestered into the core of a lipoproteinparticle-> HDL) -> increased cholesterol to phospholipid ratio, producing an absolute increase in surface area of RBC membranes |
|
|
Where is folate absorbed?
|
Jejunum |
|
|
Glossitis a/w? |
Folate deficiency and Vitamin B12 deficiency b/c cells can not divide properly b/c these are high turnover cells that require substantial nucleic acid synthesis that relies on Folate being available. Vit B12 contributes y removing CH3 from THF and transferring it to Homocysteine |
|
|
what kind of leukocyte is characteristic of macrocytic anemias? |
Hypersegmented neutrophils |
|
|
Methylmalonic Acid usefulness as a lab test? |
Differentiates Vit B12 deficiency vs. Folate deficiency
MMA Normal in Folate Deficiency b/c conversion of MMA -> Succinyl CoA relies on Vit B12 and this is not affected in folate deficiency |
|
|
Where is intrinsic factor made? Where are they located?
|
gastric Parietal cells in body of stomach |
|
|
Describe path of Vitamin B12 Absorption |
1) Dietary form is bound to animal protein which is then cleaved by amylase from salivary glands
2) Salivary gland also secrete R-binder which stabilized Vit B12 until it reaches stomach
3) In Small Intestine, pancreastic proteases cleave R-binder
4) Vit B12 then binds IF from gastric parietal cells & together they are absorbed
4) IF-B12 complex then absorbed in ileum. |
|
|
Where is B12-IF complex absorbed? |
Ileum |
|
|
pernicious anemia arises from? |
autoimmune destruction of parietal cells, which can now not produce IF necessary for Vit B12 absorption
This is called Pernicious, b/c it takes a long time for Vit B12 stores to be exhausted |
|
|
Causes of vitamin B12 deficiency? |
1) Pancreatic insufficiency - inability to cleave Vit B12-R-binder complex |
|
|
deficiency in vegans |
Vitamin B12
|
|
|
What causes Subacute combined degeneration of spinal cord. What is the presentation? |
Vitamin B12 deficiency-> Buildup of Methylmalonic Acid in Oligodendrocytes of Spinal Cord causing damage (B/C Vit B12 is a necessary co-factor in conversion of Methylmalonic Acid to Succinyl CoA) |
|
|
Spastic Paresis occurs due to damage where in the spinal cord? |
lateral corticospinal tract (CorticoMotor pathway) |
|
|
proprioception and vibratory sensation dysfunction is due to damage where in the spinal cord? |
dorsal column (DCML-Dorsal Column Medial Lemniscus Pathway) |
|
|
What two reactions require Vitamin B12 |
1) Conversion of Methylmalonic Acid to Succinyl CoA which is important for fatty acid synthesis (This is why Vit B12 deficiency leads to neuropathy-b/c MMA builds up/leads to damage in oligodendrocytes of spinal cord)
|
|
|
Reasons for Folate Deficiency |
1) Dietary Deficiency due to poor diet- Elderly, Alcoholics
2) Increased Demand - Hemolytic Anemia, Pregnancy, Cancer
3) Methotrexate - Competitively Inhibits dihydrofolate reductase (DHFR- enzyme necessary for THF synthesis), with a one thousand-fold greater affinity for DHFR than that of folate
DHFR catalyses the conversion of dihydrofolate to the active tetrahydrofolate
B/C Folic acid is needed for the de novo synthesis of the nucleoside thymidine, and is essential for purine and pyrimidine base biosynthesis, Methotrexate, therefore, inhibits the synthesis of DNA, RNA, thymidylates, and proteins |
|
|
Lab findings of Vit B12 deficiency |
↓ Vit B12 |
|
|
The P's of Parietal Cells |
1) Proton Pumps
2) pH is lowered b/c secrete H+ (acid) 3) Pernicious Anemia - Autoimmune destruction of Parietal Cells b/c IF no longer produced-> Vit B12 def.
4) Pink - compared to more bluish Chief Cells |
|
|
MCV in Microcytic Anemia
Normal MCV Range
MCV in Macrocytic Anemia |
< 80
80-100
> 100 |
|
|
Describe reticulocyte |
Slightly larger than normal RBC with a bluish hue due to residual RNA from protein synthesis |
|
|
Normocytic Anemia |
No deficiency in the lack of RBC components, therefore the RBC's are of Normal size, but the Anemia is due to
1) Destruction of RBC's outside of BM (Retic Count should Incr) 2) Decreased production in BM (Retic Count will be low) |
|
|
Reticulocyte correction in anemia |

Nml. Reticulocyte Percentage in Anemia should be >3% (this suggests that BM is responding properly to anemia by generating new RBC's). However, b/c in cases of anemia %Reticulocyte can be artificially high b/c of low RBC's (e.g. 2/100 now 2/50 ->the number of Reticulocytes has NOT Incr., but the % has artificially)
For this Reason, we take the Retic% and multiply it by (Hematocrit/45). B/c Nml. Hematocrit is 45, and in Anemia Hematocrit (or % of Blood VOlume taken by RBC's) will be low, therefore correcting the calculation |
|
|
Types of Hemolysis |
Intravascular Hemolysis - inside Blood Vessels
Extravascular Hemolysis - RBC's destroyed in Reticular Endothelial System by Macrophages in the Spleen/Liver/Lymph nodes
In both, Reticulocyte COunt will Incr., b/c BM functioning properly |
|
|
Describe digestion of RBC's by macrophages |
Macrophages of RES (Reticular Endothelial System digest RBC's and breakdown their components)
Globin --> Amino Acids
unconjugated bilirubin binds albumin (b/c fat soluble) and is delivered to liver
In liver, unconjugated bilirubin is conjugated and excreted into bile |
|
|
Clinical presentation of extravascular hemolysis |
1) Anemia b/c RBC's are being destroyed in RES (Reticular Endothelial System)
2) Splenomegaly (hypertrophy due to excess digestion of RBC's by Macrophages)
|
|
|
Lab findings for intravascular hemolysis |
RBC's lysed and Hb pours out into blood. The small amounts of Haptoglobinin blood bind up some of the Hb and complex with it-> low levels of free floating Haptoglobin. The rest of Hb accumulates in blood->Hemoglobinemia. It will ultimately make its way to kidneys/urine -> Hemoglobinuria.
-- Hemoglobinuria |
|
|
Most common protein defects causing hereditary spherocytosis |
spectrin, ankyrin, band 3.1 - Anchoring proteins connecting RBC cytoskeleton to its PM (Plasma Membrane)
These Blebs are removed by MPhages, leading to loss of membrane.
B/c cytoplasm stays normal, but PM is progressively disappearing, the RBC loses its biconcave disks shape, in favor of a sphere.
These spherocytes, also become smaller with time as blebs form/removed by Mphages.
These Spherocytic RBC's function normally but lose their flexibility/ability to maneuver within the spleen with time as they move through the RES (Reticular Endothelial System) -> they get stuck and are destroyed by Macrophages. The spleen will hypertrophy due to hypertrophy of Macrophages digesting these RBC's (splenomegaly) |
|
|
Describe RDW in Hereditary spherocytosis |
SINCE the cause of the spherocytosis is destruction of blebs of membrane from the RBC's by the spleen, this means that older cells will be smaller due to loss of membrane compared with newer cells -> increased variability in RBC size |
|
|
What is MCHC? How does it change in hereditary spherocytosis? |
Mean Corpuscular Hemoglobin Concentration. It is INCREASED in hereditary spherocytosis due to the relative increase in [Hb] per size of RBC since the spleen is picking off blebs of the RBC making RBC volume smaller, but Hb amount stays constant |
|
|
Clinical features of Hereditary Spherocytosis |
1) Splenomegaly (increased RBC destruction results in hypertrophy of splenic Macrophages),
2) Jaundice with unconjugated bilirubin (b/c amount of unconjugated bilirubin overwhelms processing ability of liver),
3) Increased risk for bilirubin gallstones-> b/c liver makes so much conjugated bilirubin |
|
|
How do you diagnose w? |
Osmotic fragility test - place RBC's in osmotic solution
|
|
|
What is the Tx for Hereditary Spherocytosis? |
The Tx is Splenectomy b/c
The problem is not having spherocytes, but rather that they get stuck in Spleen and get destroyed by Macrophages Removing Spleen, allows Spherocytes to survive |
|
|
What kind of cell will you see on peripheral blood smear post-splenectomy? |
Howell-Jolly bodies (fragments of nuclear material in RBCs). |
|
|
Profile of Hereditary Spherocytosis post-splenectomy |
1) Spherocytes (b/c not destroyed in spleen) 2) Howell-Jolly Bodies (b/c not removed in spleen) 3) Anemia resolves - b/c Spherocytes not destroyed |
|
|
Sickle Cell Anemia protects against? |
protects against Plasmodium Falciparum
|
|
|
What drug will increase levels of HbF?
|
hydroxyurea, MOA unknown
|
|
|
HbS |
when both Beta alleles have substitution of Valine (non-polar) in place of glutamic acid (polar) ->Resulting in hydrophobic polymerization when this HbS gives up O2 |
|
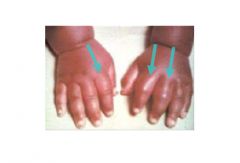
How do HbS cells sickle, what happens? |
HbS polymerizes (NOT Covalent bonds. It is reversible) in response to loss of O2 induced by: 1) Hypoxemia 2) Dehydration 3) Acidosis
This HbS polymerization causes the RBC's to become needle-like/sickle shaped
RBC's Sickle and Unsickle repeatedly depending on [O2]. Repeated sickling/unsickling -> membrane damage (due to HbS polymer damaging PM) -> Hemolysis |
|
|
Clinical manifestations of Sickle Cell Disease |
A result of RBC membrane damage. Leads to...
3) Target Cells seen in Sickle Cell due to dehydration of RBC's as they get damaged (air out of basketball) |
|
|
Seen in both Extravascular & Intravascular Hemolysis due to Sickle Cell Anemia |
Massive Erythroid Hyperplasia due to Anemia -> 1) Expansion of hematopoeisis to Skull & Facial bones-> Crew cut appearance on X-ray/Chipmunk faces in response
2) Hepatomegaly - due to Extramedullary Hematopoesis (Normally in Liver/Spleen, but here only Liver b/c Spleen destroyed)- this is part of expansion of hematopoesis due to anemia
3) Risk of Aplastic Crisis w/ Parvovirus B19 |
|
|
Tx for Sickle Cell Anemia |
Hydroxyurea to elevate HbF |
|
|
When does Sickle Cell Anemia first present? |
6 months after birth b/c for the first 6 months levels of HbF are still pretty high, therefore RBC'S are not likely to sickle |
|
|
Extravascular Hemolysis can be caused by? |
1) Hereditary Spherocytosis
2) Sickle Cell Anemia |
|
|
Haptoglobin |
Circulates in serum and scavanges Hb in intravascular hemolysis |
|
|
Irreversible Sickling leads to... |
Vaso-occlusion of Sickled RBC's leading to Infarction of Tissues |
|
|
Consequences of Vaso-occlusion in Sickle Cell Anemia |
1) Dactylitis - Swollen hands and feet due to vaso-occlusive infarcts of bones - Common presenting sign in children at >6 m.o.
2) Autosplenectomy - Infarction of Spleen leading to shrunkedn/fibrotic spleen due to Vaso-occlusive Crisis
|
|
|
Consequences of Autosplenectomy? |
1) Increased risk of infection w/ encapsulated organisms (MCC of death in children) - B/c most Plasma cells secrete Ab in Spleen, but will now be missing (ex. H. Influenza, Strep Pneumo,..therefore you must vaccinate sickle cell child against these organisms)
2) Osteomyelitis MC (most commonly) due to Salmonella paratyphi (Normally MCC of osteomyelitis is Staph Aureus)
3) Howell-Jolly bodies on blood mear b/c not removed by splenic Macrophages |
|
|
Most common cause of death in adults with sickle cell disease? |
Acute Chest Syndrome -- vasoocclusion in pulmonary microcirculation. Presents with chest pain, shortness of breath and lung infiltrates. Can be precipitated by pneumonia (pneumonia infection->inflamamtion->vasodilation->slows RBC transit time->slow moving RBC's give up more O2 from Hb due to hypoxia/vasodilation->sickling->occlusion) |
|
|
Acute Chest Syndrome |
Usually follows pneumonia b/c
Inflammation-> Vasodilation -> slowed RBC transit-> More time for HbS to release O2 in hypoxic/acidemia->sickling of RBC's->Vaso-occlusion in pulmonary microcirculation-> Chest PAIN!!!, SOB, lung infiltrates
|
|
|
Vaso-occlusion in Sickle Cell Anemia causing pain in various organs |
Pain Crisis |
|
|
Renal Papillary Necrosis |
Hematuria & proteinurea due to vaso-occlusion in kidney blood vessels due to Sickle Cell Disease |
|
|
Sickle cell train - clinical presnetation |
Heterozygous pt. makes 45% HbS & 55% HbA (b/c production of functional alpha2Beta2 is more efficient). Since >50% HbS is necessary for an RBC to sickle, these pts generally do not exhibit sickling, with 1 Exception |
|
|
What organ is most susceptible to damage in sickle cell TRAIT individuals? What is the classic finding of long term complications related to this fact? |
renal medulla- b/c highly hypoxic (due to little blood flow) & hypertonic -> leading to Sickling ->leading to vaso-occlusion in medullary Vasa Recta-> microinfarctions->microscopic hematuria->Eventually Decreased ability to concentrate urine |
|
|
What chemical induces sickling even if small amount of HbS? |
Metabisulfite |
|
|
Name the types of hemoglobin present in sickle cell disease and trait |
Sickle Cell Disease - HbS= 90%, HbF=8% HbA2 = 2% HbA= 0% (No HbA present!!) HbA = 55% HbS = 43% HbA2 = 2% |
|
|
Hemoglobin C high yield association? |
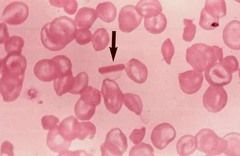
HbC Crystals seen on blood smear- b/c Beta chain of HbC has LySIne in place of glutamic acid->mild anemia due to Extravascular Hemolysis |
|
|
Paroxysmal Nocturnal Hemoglobinurea (PNH) |
ACQUIRED defect (mutaiton w/ age) in myeloid stem cell in the gene for GPI (GPI anchors DAF (Decay Accelerating Factor= accelerates decay of C3 Convertase) and MIRL (Membrane INhibitor of Reactive Lysis) to RBC) -> DAF and MIRL are not present on RBC (DAF & MIRL protect RBC from complement in the blood)-> complement can lyse the RBC's |
|
|
What cells will be missing GPI in PNH? ( Paroxysmal = sudden recurrence or onset of symptoms) |
All descendentsts of myeloid precursor w/ acquired mutation in GPI - RBC's Granulocytes, Platelets-> No DAF/MIRL->these cells are suceptible to complement destruction-> on some nights (paroxysmal) they have RBC destruction-> Dark urine due to Hb in Urine-> Additionally Platelets & Granulocytes are lysed |
|
|
Intravascular Hemolysis |
Hemoglobinemia Hemoglobinuria Hemosiderinuria - days after hemolysis b/c some Hb absorbed by PCT cells, they accumulate Fe (siderin) and when they slogh off, hte hemosiderin ends up in urinecreen S |
|
|
Screen for PNH |
Sucrose test-> Sucrose activates compliment in pts serum sample -> hemolysis
Confirmatory Test - Acidified Serum Test - Acidification activates Compliment
New Confirmatory Test - Flow Cytometry detects absence of CD55 (DAF) -> confrims absence of GPI |
|
|
Main Cause of Death in PNH |
Thrombosis in Cerebral, hepatic, portal veins b/c lysis of platelets causes fragments of platelets to activate Coagulation Cascade |
|
|
Complications of PNH |
Fe deficiency Anemia - due to loss of Fe w/ Hbinuria
AML in 10% of pts, simply b/c if PNH pts have mutation in GPI, they are likely to have other mutations that can lead to Leukemia |
|
|
Glutathione Reductase Pathway |
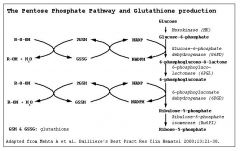
G6PD reduces oxidized glutothione so that it can continue to reduce H2O2 that would normally cause oxidative damage to RBC |
|
|
Variants of G6PD?
|
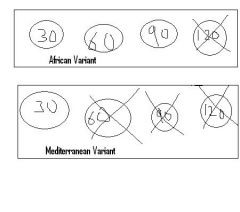
African variant -- mildly reduced t1/2 of G6PD leading to mild intravascular hemolysis with oxidative stress (b/c only very old RBC's lose G6PD and lyse) |
|
|
What does oxidative stress do to Hb? |
Precipitates Hb as Heinz bodies |
|
|
Causes of Oxidative stress? |
Infections, Drugs (sulfa, nitryl, primaquine), and Fava beans (Mediterranean diet) |
|
|
Classic sign in G6PD? |
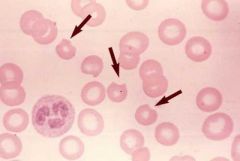
Heinz bodies -- precipitated Hb due to oxidative stress. These precipitates are bitten out of the RBC in the spleen leading to 'bite cells' |
|
|
G6PD Clinical signs?
|
Intravascular hemolysis causing hematuria. This causes back pain since Hb is nephrotoxic in PCT. Hemoglobinuria seen w/in hours of oxidative stress |
|
|
Normocytic anemias with Predominantly Intravascular hemolysis |
PNH, G6PD, IHA(Immune Hemolytic Anemia), Microangiopathic Hemolytic Anemia, Malaria |
|
|
Heinz Preparation?
|
Test for G6PD |
|
|
Immune Hemolytic Anemia |
IgG or IgM mediated destruction of RBC's |
|
|
IgG type Immune Hemolytic Anemia Clinical Signs |
IgG will present with spherocytes since antibody coating of RBC will cause splenic Macrophages to pick off bits and pieces of cytoplasm->Spherocytes
IgG mediated is therefore ->Extravascular Hemolysis b/c Spherocytes will get stuck/destroyed in Spleen with time |
|
|
IgG type Immune Hemolytic Anemia associated with...? |
extravascular hemolysis |
|
|
Treatment of IgG type Immune Hemolytic Anemia |
1) Cessation of offending Drug if it exists
2) Steroids
3) IVIG -- eat IVIG instead of RBCs
4) Splenectomy - (Last Resort) removes source of Ab & source of RBC destruction (Splenic Macrophages) |
|
|
IgM type Immune Hemolytic Anemia Associations |
INTRAvascular hemolysis due to IgM binding in relative cold temp of extremities. A/w Mycoplasma pneumonia (cold agluttinins) that cause complement activation->MAC Complex formation->Intravascular Hemolysis
|
|
|
Overview of Coombs Test |
Done to identify IHA Direct Coombs Test- Confirms presnce of Ab coated RBC's --- Do I have RBC's already bound to IgG? |
|
|
Microangiopathic Hemolytic Anemia |
Formation of Schistocytes due to creation of thrombi in microcirculation. |
|
|
'helmet' shaped RBC
|
Schistocyte |
|
|
Malaria transmitted by?
|
Anopheles mosquito transmits the Plasmodium species of bug, infects RBC's & Hepatocytes |
|
|
Plasmodium falciparum vs vivax/ovale? |
Plasmodium falciparum -- daily fever
Fever happens every time RBC's rupture
RBC rupture happens every time Plasmodium reproduces & starts new life cycle |
|
|
Intravascular Hemolysis can be caused by? |
G6PD |
|
|
Extravascular Hemolysis in Malaria |
A little bit b/c some RBC's are destroyed by Splenic Macrophages ->Hypertrophy of these Macropahges causes Splenomegaly |
|
|
Anemia's Due to Underproduction |
Decreased production of RBC's by defective BM
Lab findings - Anemia w/ low Retic COunt b/c BM not capable of compensating for low RBC levels |
|
|
Causes of BM UNderproduction |
1) Anything that would cause Macrocytic or Microcytic anemia b/c low Fe or B12/ Folate deficiency will also interfere w/ Reticulocyte production
2) Renal failure - b/c EPO will be low and EPO extends RBC precursor lifespan
3)Damage to BM precursor cells (e.g. Parvovirus B19) |
|
|
Parvovirus B19 effect on Progenitor Red cells |
1) Temporarily halts Erythropoeisis b/c infects progenitor Red cells 2)Significant Anemia in pts already struggling to produce enough RBC's - in healthy pts it is not a problem b/c enough RBC reserves 3)Infection is self-limited - resolves in 7-10 days |
|
|
Aplastic Anemia |
Damage to HSC Results in Pancytopenia (Platelets/RBC's/WBC LOWV) w/ low Retic Count |
|
|
Causes of Aplastic Anemia |
Drugs or Chemicals Viral INFECTION AUTOIMMUNE DAMAGE |
|
|
Aplastic Anemia Histology |
ADIPOSE TISSUE REPLACES HEMATOPOEITIC ELEMENTS IN BM |
|
|
Tx of Aplastic Anemia |
1) Cessation of Causative Dx 2) Transfusions 3) Marrow Stimulating Factors (EPO for RBC's, GM-CSF & G-CSF for Granulocytes) of whatever HSC's remain 4) Immunosupression b/c one of Etiolgies of Aplastic Anemia is Auto-immune 5) BMT (last resort) |
|
|
Myeophthisic Process |
Pathologic Process that replaces BM-> Hematopoesis is impaired->pancytopenia
e.g. metastatic cancer replacing BM would interfere w/ Hematopoesis->pancytopenia |