Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
44 Cards in this Set
- Front
- Back
|
Adrenal cortex has three layers each secreting a different hormone
-Glomerulosa -Fasciculata -Reticularis - |
-Mineralocorticoids (aldosterone)
-Glucocorticoids (cortisol) -sex steroids (testosterone) |
|
|
What is congenital adrenal hyperplasia?
|
Excess of steroids with hyperplasia of both adrenal glands. Inherited 21 hydroxylase deficiency is the most common cause
|
|
|
How does adrenal hyperplasia develop?
|
Due to 21 hydroxylase deficiency, steroidogenesis is shunted toward sex steroid production. Deficiency of cortisol leads to increased ACTHC secretion (lack of negative feedback) leading to bilateral adrenal hyperplasia.
|
|
|
What are the findings of congenital adrenal hyperplasia?
|
salt wasting with hyponatremia, hyperkalemia and hypovolemia due to lack of aldosterone.
Life threatening hypotension Enlarged clitoris in females Precocious puberty in males |
|
|
What can cause primary acute adrenocortical insufficiency?
|
-Sudden increase in glucocorticoid requirement in patients with adrenal crisis (chronic insufficiency)
-Rapid withdrawal of steroids, or failure to increase steroid doses in periods of stress in patientes with adrenal suppression secondary to long term glucocorticoid therapy. -Massive adrenal hemorrhage (waterhouse friderichsen syndrome) |
|
|
Whats the waterhouse friderichsen syndrome?
|
-Overwhelming septicemic infection due to meningoccocus
-Rapidly progressive hypotension and shock -DIC purpura -Massive adrenal hemorrhage with adrenal insufficiency |
|
|
What can cause primary chronic adrenocortical insufficiency "Addison disease?
|
Sudden cessation of glucocorticoid therapy
Autoimmune destruction (autoimmune adrenalitis) Infection (TB, Histoplasmosis) Hypopituitarism |
|
|
What are the symptoms of addison's disease?
|
Weak, weight loss, anorexia, hypotension, N+V
Hyperpigmentation (proopiomelanocortin) if primary Hypoglycemia, hyperkalemia if primary |
|
|
Autoimmune adrenalitis involves
|
Autoimmune polyendocrinopathy syndrome type 1. and autoimmune polyendocrinopathy syndrome type 2.
|
|
|
What causes Autoimmune polyendocrinopathy syndrome type 1?
|
AIRE regulator absence. This thymic transcription factor drives the expression of peripheral tissue antigens so that self reactive t cells undergo clonal deletion. In absence of AIRE, autoimmune attack develops
|
|
|
What is the clinical presentation of Autoimmune polyendocrinopathy syndrome type 1?
|
Chronic mucocutaneous candidiasis and abnormalities of the skin, dental enamel and nails ocurring in association to other autoimmune disorders
|
|
|
What are the clinical manifestations of Autoimmune polyendocrinopathy syndrome type 2?
|
Presents in early adulthood as a combination of adrenal insufficiency and autoimmune thyroiditis or type 1 diabetes. mucocutaneous candidiasis and abnormalities of the skin do not occur.
|
|
|
What causes secondary adrenacortical insufficiency?
|
Occurs with any hypothalamic or pitutitary disorder leading to a diminished ACTH production (tumor, infection, infarction)
-It can an isolated deficiency or associated with decreased levels of other pituitary hormones |
|
|
How is secondary adrenocortical insufficiency distinguished from primary adrenocortical insufficiency (Addison disease)
|
Absence of hyperpigmentation
Near-normal aldosterone levels since production is largely independent of ACTH; thus hyponatremia and hyperkalemia are not feats of secondary adrenal insufficiency |
|
|
What is hypercortisolism and what causes it?
|
It is an excess of cortisol (Cushing syndrome)
It is caused by ACTH Dependent and ACTH Independent reasons |
|
|
Cushing syndrome ACTH Dependent etiologies
|
Pituitary tumor: ACTH secreting tumor mostly in women.
ACTH suppressible by high dose dexamethasone Ectopic ACTH: mostly in men 40-60 Small cell CA lung Carcinoid Not suppressible |
|
|
Cushing syndrome ACTH independent causes
|
-Exogenous steroids: bilateral adrenal atrophy, steroids supress ACTH secretion (negative feedback)
-Adenoma -Carcinoma -Micronodular hyperplasia -Macronodular hyperplasia |
|
|
What are the clinical manifestations of cushing disease?
|
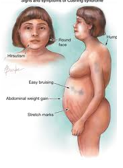
Muscle weakness, moon facies, buffalo bump and truncal obesity. Abdominal striae, HTN, osteoporosis
|
|
|
What is conn syndrome?
|
It is excess aldosterone due to adrenal adenoma most commonly, and sporadic adrenal hyperplasia and adrenal carcinoma less commonly
|
|
|
Cushing’s Syndrome: Diagnostic Tests
Establish GC Excess, any 2 of: |
-Salivary cortisol, late night (x 2)
-Elevated 24h urine free cortisol (x 2) If > 3x normal = Cushing’s syndrome Best screen (have creat also) -Overnight dexamethasone suppression 1 mg at MN –> AM cortisol Serum cortisol < 1.8 µg/dl r/o Cushing’s F(+); obesity, depression -2–day dexamethasone suppression 0.5 mg q 6h x 8 Serum cortisol < 1.8 µg/dl r/o Cushing’s syndrome Urine cortisol < 10 µg/d r/o Cushing’s syndrome May also be used to determine source Cushing’s DISEASE will suppress |
|
|
Cushing’s Syndrome Establish Source (Serum ACTH)
|
-Normal or high
Pituitary or ectopic source –> MRI pituitary Tumor –> surgery No tumor –> petrosal v. sampling ACTH high –> surgery ACTH not high –> CT chest -Low Adrenal source of GC CT/MRI adrenals |
|
|
What is the clinical presentation of conn's syndrome?
|
Hypertension with hypokalemia
NO EDEMA Atrial natriuretic peptide, downreg Na–Cl co–transporter, pressure natriuresis |
|
|
How is Conn's syndrome diagnosed?
|
Elevated plasma aldosterone + low plasma renin
Image adrenals |
|
|
What is salt wasting syndrome?
|
Associated with a complete deficiency of 21 hydroxylase activity and thus absent aldosterone or cortisol production
|
|
|
How is salt wasting syndrome recognized?
|
shortly after birth, by salt wasting hyponatremia and hyperkalemia leading to acidosis, hypotension and cardiovascular collapse. Virilization in females
|
|
|
What is simple virilizing adrenogenital syndrome without salt wasting?
|
associated with incomplete loss of hydroxylase activity. Patients have enough aldosterone to avoid a salt wasting crisis, but reduced cortisol production still drives ACTH secretion and ultimately increased testosterone synthesis
|
|
|
Non classic (late onset) adrenal virilism
|
Partial 21 hydroxylase deficiency results in no symptoms or only subtle feats of adrogenic excess later in life (hirsuitism, acne, or menstrual irregularities)
|
|
|
What is the gross and microscopic appearance of cortical carcinomas?
|
Grossly: Tumors are variegated with areas of hemorrhage, cystic change and necrosis
Microscopically: Cells range from well differentiated to markedly anaplastic |
|
|
Can functioning tumors be distinguished morphologically?
|
no.
|
|
|
What is the gross and microscopic appearance of cortical adenomas?
|
Grossly: Well circumscribed, yellow-brown lesions up to 2.5 cm. In nonfunctioning adenomas, the adjacent cortex is normal thickness, in functioning neoplasms they adjacent cortex is atrophic
Microscopically: Cells range from well differentiated to anaplastic |
|
|
What is the incidence of pheochromocytoma?
|
Very uncommon tumors of chromaffin cells. The tumors produce catecholamines and present with htn.
|
|
|
What is the rule of 10's that pheochromocytoma follows?
|
10% for bilateral, familial, malignant, located outside adrenal medulla.
|
|
|
What are the clinical feats of pheochromocytoma?
|
Episodic HTN, headache, palpitations, tachycardia and sweating.
|
|
|
How is pheochromocytoma diagnosed?
|
By increased serum metanephrines and increased 24 hour urine metanephrines and vanillyl mandelic acid
|
|
|
The sole criterion for pheochromocytoma malignancy is
|
metastases. Microscopically it's composed of clusters of polygonal to spindle shaped chief cells (expressing S-100) all delimited by rich vascular network.
|
|
|
what is a paranglioma?
|
Extra-adrenal paragangliomas (often described as extra-adrenal pheochromocytomas) are closely related, though less common, tumors that originate in the ganglia of the sympathetic nervous system and are named based upon the primary anatomical site of origin.
|
|
|
Multiple endocrine neoplasia (MEN) type 1 is characterized by 3 Ps
|
Parathyroid: Primary Hyperpthism due to hyperplasia or adenoma is the first manifestation
Pancreas: Functional aggresive tumors. Pancreatic peptide most commonly hormone produced>Insulinomas>gastrinomas Pituitary: Prolactinomas |
|
|
MEN-1 is caused by
|
germline mutations in the MEN 1 tumor suppresor gene, encoding the protein MENIN, which is a component of several different transcription factor complexes.
|
|
|
MEN 2 is divided into 3 distinct syndromes
|
-MEN-2A (Simple syndrome)
-MEN 2-B -Familial medullary thyroid cancer |
|
|
Men 2A (Sipple syndrome) is characterized by
|
thyroid medullary carcinoma, pheochromocytoma, and parathyroid hyperplasia with hypercalcemia. It is caused by germline gain of function of RET protooncogene
|
|
|
MEN 2B is characterized by
|
thyroid medullary carcinoma and pheochromocytomas, hyperpthis does not develop. Instead patients develop neuromas or ganglioneuromas
|
|
|
How do pts wit MEN 2B present?
|
Marfanoid habitus, with long axial skeletal feats and hyperextensible joints. The syndrome is caused by a unique, single amino acid substitution in the RET leading to constitutive activation of its tyrosine kinase activity
|
|
|
Familial medullary thyroid cancer is a variant of MEN 2A with a strong predisposition to
|
thyroid malignancy but without other clinical manifestations.
|
|
|
Genetic screening is life saving for at risk family members of patients with
|
MEN 2 syndromes because thyroidectomy can potentially mitigate the fatal complications of medullary carcinoma
|