Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
29 Cards in this Set
- Front
- Back
|
Small, discrete cortical tumors
Arise from the renal tubular epithelium Most frequently papillary |
Renal adenoma
|
|
|
Renal adenoma morphology
|
Complex, branching, papillomatous structures with numerous complex fronds
|
|
|
Present in 25% to 50% of patients with tuberous sclerosis, a disease characterized by
Lesions of the cerebral cortex Skin abnormalities |
Angiomyolipoma
Benign tumor consisting of Vessels Smooth muscle Fat |
|
|
Epithelial tumor Tan or mahogany brown with a central scar
|
Oncocytoma. Arise from the intercalated cells of collecting ducts
|
|
|
Oncocytoma are composed of
|
-Composed of large, eosinophilic cells having small, round, benign-appearing nuclei that have large nucleoli
-, the eosinophilic cells have numerous mitochondria -Familial cases may be multicentric |
|
|
Generally 0.5 cm in size or less
Spindle cells in loose stroma with entrapped tubules |
Medullary Fibroma. Incidental finding also called a renomedullary interstitial cell tumor
Not associated with any renal diseases |
|
|
Hematuria with palpable mass, flank pain, long standing fever and associated with paraneoplastic syndromes
|
Renal cell carcinoma (adenocarcinoma of the kidney) which arises from tubular epithelium
Associated with von Hippel Lindau disease. Most common in men in their 60s and it is the most common renal malignancy |
|
|
Main risk factors for renal cell carcinoma
|
Tobacco is the most significant risk factor (double)
Obesity (particularly in women) Hypertension Unopposed estrogen therapy Exposure to asbestos, petroleum products, and heavy metals Increased incidence in patients with chronic renal failure |
|
|
Von Hippel-Lindau (VHL) syndrome
|
hemangioblastomas of the cerebellum and retina
renal cysts bilateral, often multiple, renal cell carcinomas (nearly all, if they live long enough) current studies implicate the VHL gene in the development of both familial and sporadic clear cell tumors. |
|
|
Renal Cell Carcinoma most cases are due to
|
sporadic reasons. 98% of these tumors, whether familial, sporadic, or associated with VHL, there is loss of sequences on the short arm of chromosome 3
VHL gene acts as a tumor-suppressor gene |
|
|
Papillary carcinoma sporadic form genetic bases
|
Trisomies 7, 16, and 17 and loss of Y in male patients
|
|
|
Papillary carcinoma familial genetic bases
|
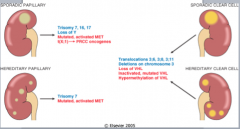
The gene for the familial form has been mapped to a locus on chromosome 7, encompassing the locus for MET
a protooncogene tyrosine kinase receptor for hepatocyte growth factor. mutated in a proportion of the sporadic cases hepatocyte growth factor (also called scatter factor) mediates growth, cell mobility, invasion |
|
|
grow from intercalated cells of collecting ducts
excellent prognosis histologic distinction from oncocytoma can be difficult. |
Chromophobe renal carcinoma
|
|
|
Carcinoma of collecting duct cells in the medulla
medullary location. |
Collecting duct (Bellini duct) carcinoma
|
|
|
Where does renal cell carcinoma spread to?
|
Lungs (more than 50%)
Bones (33%) Rregional lymph nodes Liver Adrenals Brain |
|
|
Huge palpable flank mass in a young child and microscopic hematuria
|
Wilms tumor. Most common primary renal tumor of childhood
4th most common pediatric malignancy in US 2-5 years |
|
|
Wilms tumor is caused by
|
deletion of WT-1 gene (tumor suppressor) on short arm of chr 11.
|
|
|
Denys-Drash syndrome
|
90% risk of Wilms tumor
Gonadal dysgenesis Early onset nephropathy (Diffuse mesangial sclerosis) Germline abnormality of WT1 Increased risk of gonadoblastomas |
|
|
Organomegaly
Macroglossia Hemihypertrophy Omphalocele Chromosome 11 band p15.5 (WT2) Abnormality of genomic imprinting |
Beckwith-Weidemann syndrome
|
|
|
papillomas to invasive carcinomas
noticeable hematuria |
Urothelial Carcinomas of the Renal Pelvis
|
|
|
Painless hematuria; irritative voiding symptoms, palpable mass, hepatomegaly
|
Transitional cell carcinoma. Associated with smoking, exposure to aniline dyes, cyclophosphamide and phenacetin abuse.
|
|
|
Scattered small cortical and medullary cysts with hyperplastic or flattened epithelium
0.5 to 2 cm in size Cysts contain clear fluid and calcium oxalate crystals |
Acquired (dialysis-associated) cystic disease
|
|
|
Acquired (dialysis-associated) cystic disease forms due to
|
obstruction of tubules by interstitial fibrosis or by crystals
|
|
|
Autosomal dominant disease with APKD1 gene mutation on chromosome 16
|
Autosomal-dominant (adult) polycystic kidney disease (ADPKD)
Infantile form is autosomal reccesive PKHD1 maps to chromosome 6p21-23 novel protein, fibrocystin |
|
|
Whats the pathology of Autosomal-dominant (adult) polycystic kidney disease (ADPKD)?
|
Replacement of renal parenchyma bilaterally with multiple large variably size cysts
|
|
|
Infantile polycystic kidney disease (ADPKD) pathology
|
closed, small, homogenous cysts that are not in continuity with the collecting system
|
|
|
CT with multiple cysts at birth
|
Infantile polycystic kidney disease (ADPKD). Results in death shortly after birth
|
|
|
Hypertension, hematuria, and palpable renal mass. CT scan shows multiple cysts in both kidneys
|
Autosomal-dominant (adult) polycystic kidney disease (ADPKD)?
|
|
|
Autosomal-dominant (adult) polycystic kidney disease (ADPKD) is associated with
|
secondary polycythemia, polycystic liver disease, berry aneurysms and mitral valve prolapse.
|