Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
47 Cards in this Set
- Front
- Back
|
Pleiotropism
|
single gene has multiple effects
|
|
|
Genetic heterogeneity
|
different mutations have similar phenotypic changes
(16 different gene mutations cause deafness) |
|
|
Penetrance
|
fewer or no phenotypic changes are noticed in reduced penetrance
given as percentage *autosomal dominant disorders |
|
|
Variability of Expression
|
Same genetic mutation leads to different phenotypic changes in different individuals
(ex: Neurofibromatosis type I) |
|
|
Age of Onset
|
delayed in AD as compared to AR disorders
|
|
|
AD disorders
(where mutation) |
diminished or absence of proteins that:
regulate complex metab pw structural proteins (collagen, cytoskel elements) less common - gain of protein function not normal in wild-type such as toxic properties in Huntington dz |
|
|
AR disorders
(general principles) |
manifest in homozygous state
complete penetrance common onset frequently early in life consanguineous marriage possible if gene has low frequency in population *enzyme usually affected in carriers, 50% of enzyme normal - usually adequate for normal function includes almost all inborn errors of metabolism |
|
|
X-linked dominant disorder example?
|
Vitamin D- resistant rickets
|
|
|
4 classifications of single gene disorders
|
Enzyme defects and their consequences
Defects in membrane receptors and transport systems Alteration in the structure, function or quantity of non-enzyme proteins Mutations resulting in unusual reactions to drugs |
|
|
Disorders associated with defects in structural proteins
(name) |
Marfan syndrome
Ehlers-Danlos Syndrome |
|
|
Marfan syndrome
|
usually missense mutation of Fibrillin-1 (FBN-1) on chromosome 15
most AD, others spontaneous abnormal fibrillin disrupts normal microfibrils in *dominant negative* manner Phenotype: tall body long extremities long fingers (arachnodactyly lax joint ligaments in hand/feet long head w/ bossing fo frontal eminences and prominent supraorbital ridges spinal deformities (kyphosis, scoliosis, slipping lumbar vertebrae) chest deformities (pectus excavatum or pigeon breast deform) ectopic lentis (subluxation and dislocation of ocular lens) cardiac (mitral valve prolapse, dilation of aortic root with secondary valve incompetence and tendency for aortic dissections which is common cause of death 30-45%) Variability seen - possibly due to heterogeneity of mutations of FBN-1 |
|
|
Ehlers-Danlos syndrome
|
Heterogenic group of disorders
**ALL secondary to defects in synthesis or structure of fibrillar collagen** AD, AR, X-linked AR associated with enzyme defects (lysyl-hydroxylase and procollagen N-peptidase) Joint hypermobility (contortionists) skin laxity joint dislocations fragile skin with poor healing scoliosis |
|
|
Disorders associated with defects in receptor proteins
(name) |
Familial hypercholesterolemia
|
|
|
Familial hypercholesterolemia
|
disorder secondary to defects in LDL receptor (mutation of receptor gene)
*most frequent mendelian disorder (1 in 500) AD type 2X to 3X increase in serum cholesterol leading to: xanthomas and premature atherosclerosis homozygotes - even higher cholesterol MI possible b4 age 20 Normal role of LDL receptors: on liver cells they snag LDL molecules and allow entry to cell LDL is degraded in lysosomes causing cholesterol release this depresses hepatocyte chol production, activates storage of chol as esters, and suppresses LDL receptor production to prevent accum of chol in hepatocytes If process disrupted, serum LDL levels increase causing xanthomas and atherosclerosis |
|
|
Familial hypercholesterolemia
(5 forms) |
Receptor defects are heterogenous w/ 5 forms:
Altered synthesis of receptor proteins (null allele) altered transport of receptors (abnormal forms accum in ER, common) altered LDL binding to receptor (receptors reach surface but fail to bind LDL or do so poorly) receptors fail to cluster in coated pits (fail to internalize bound LDL) receptors fail to release LDL (degraded by lysosome and can't be recycled) |
|
|
Disorders associated with defects in enzymes
(name) |
Lysosomal Storage Disorders:
Tay-Sachs Disease Niemann-Pick Disease Gaucher Disease Mucopolysaccharidoses Glycogen storage disorders Alkaptonuria |
|
|
Disorders associated with defects in proteins that regulate cell growth
|
Neurofibromatosis
|
|
|
Lysosomal Storage Disorders
(general characteristics, 5 mechanisms) |
lysosomes contain acid hydroxylases that are "tagged" with terminal mannose-6-phosphate groups in Golgi
This addresses them as they bind internal membrane receptors in Golgi and pinch off to form small vesicles that fuse with lysosomes Lysosomes and their acid hydroxylases degrade large molecules derived from internal organelles (autophagy) or obtained from outside of the cell by phagocytosis (heterophagy) Failure of lysosomes to fully degrade large molecules leads to accumulations that cause lysosomal storage disorders 5 mechanisms: synthesis of inactive enzymes defects in post-translational processing lack of enzyme activator or protector proteins lack of substrate activator proteins lack of transport proteins to egress the digested lysosomal materials |
|
|
Tay-Sachs Disease
|
(GM2 Gangliosidosis: Hexosaminidase alpha-subunit defiency)
AR mutation of alpha-subunit of hexosaminidase on chromosome 15 prevalent among Ashkenazic Jews (1 in 30) Clinical: involves neurons and retina neurons distended by ganglioside filled lysosomes (onion skin appearance on EM) neurons eventually destroyed and phagocytes accumulate the complex lipids **cherry red macula is prominent** begin at 6 mo with progressive motor and mental deterioration with muscle flaccidity, blindness, and progressive veg state and death by 2-3 yrs (other GM2 gangliosidoses include Sandhoff dz (beta-subunit defect) and activator deficiency, both with similar clinical picture) |
|
|
Niemann-Pick Disease: Type A
|
AR, common in Ashkenazic Jews
missense mutation of sphingomyelin protein and almost complete lack of the enzyme sphingomyelin accumulates particularly in mononuclear phagocytic cells, which have a foamy cytoplasm and are Oil Red O positive on frozen sections Concentric laminated structures seen on EM enlarged: spleen liver lymph nodes bone marrow tonsils GI tract lungs CNS: small gyri, large sulci with vacuolated neurons cherry red macula in 1/3 can manifest at birth with abdominal growth secondary to hepatosplenomegaly Progressive failure to thrive with vomiting, fevers, lymphadenopathy, and deterioration Death at 1-3 yr |
|
|
Niemann-Pick Disease: Type B
|
AR, common in Ashkenazic Jews
Less severe course than type A generally no CNS involvement Organomegaly with survival into adulthood |
|
|
Niemann-Pick Disease: Type C
|
AR
NPC-1 mutation defects in cholesterol trafficking: chol accumulates in affected cells CNS: astrocytes prominently affected and degeneration of terminal axons and dendrites clinically heterogenous with: some hydrops fetalis and stillbirths neonatal hepatitis and chronic forms (progressive neurologic damage) most present in childhood with: ataxia supranuclear palsies psychomotor regression hepatosplenomegaly dysarthria |
|
|
Gaucher Disease
|
AR cluster of disorders with mutation in glucocerebrosidase gene
**Most common of lysosomal storage disorders** results in accumulation of glucocerebrosides in phagocytic cells of the body 3 clinical subtypes: ***Type I - 99% chronic non-neuropathic form splenomegaly and skeletal changes common in Jews from Europe reduced but detect levels of glucucerebrosidase with some shortening of longevity Type II (acute neuropathic Gaucher disease) no detect levels of glucocerebrosidase no predilection for Jews progressive CNS degeneration with hepatosplenomegaly and death at early age Type III: intermediate form systemic involvement like Type I with progressive CNS changes beginning in teens to twenties *All characterized by the Gaucher cell: disteneded phagocytic cells with a "crumpled tissue paper" appearance of cytoplasm Detect homozygotes by leukocytes level of the enzyme recombo therapy successful in Type I patients |
|
|
Mucopolysaccharidoses
|
closely related group of disorders secondary to abnormalities in enzymes that degrade mucopolysacchcarides
Listed as MPS 1-VII ***All are AR, except Hunter syndrome which is X-linked recessive (Males are hunters)*** Generally progressive w/ multiple organs involved: liver, spleen, heart, skeletal system, blood vessels Most have course facial features, clouding of cornea, joint stiffness, mental retardation Phagocytic cells have clear cytoplasm - PAS positive, finely granular MPS 1-Hurler has abnormal alpha-1-iduronidase, death by 6-10, secondary to cardiovascular complications MPS II - Hunter syndrome has abnormal iduronate-2-sulfatase and a milder clinical picture with corneal clouding |
|
|
Glycogen storage disorders
(in general) |
defects in synthesis and catabolism of glycogen
may be limited to a few tissues or be systemic 3 major groups: Hepatic forms lead to hepatomegaly and hypoglycemia Myopathic forms lead to muscle cramps and no elevation of exercise induced lactate levels Forms associated with lack of acid maltase and branching enzyme cause storage abnormalities in many organs and early death |
|
|
Glycogen storage disorders
Hepatic forms |
Hepatic forms lead to hepatomegaly and hypoglycemia:
von Gierke - lack glucose-6-phosphatase - Type I lack of liver phosphorylase - Type VIII lack of debranching enzyme - Type III |
|
|
Glycogen storage disorders
Myopathic forms |
lead to muscle cramps and no elevation of exercise-induced lactate levels
McArdle disease (lack of muscle phosphorylase), Type V lack of muscle phosphofructokinase - Type VII |
|
|
Glycogen storage disorders
forms associated with lack of acid maltase and branching enzyme |
cause glycogen storage abnormalities in many organs and are associated with early death:
Pompe disease - lack of acid maltase, Type II - prominent cardiomegaly lack of branching enzyme, Type IV |
|
|
Alkaptonuria (onchronosis)
|
first human inborn error of metab discovered
AR lack of homogentisic oxidase: blocks phenylalanine metab at the level of homogentisic acid causes deposition in soft tissue with black discoloration, particularly over ears, nose, and cheecks deposition in articular cartilage can lead to joint injury excreted in urine, which turns black upon standing Chromosome 3q21 |
|
|
Neurofibromatosis Type I
|
(von Recklinghausen dz)
AD, secondary to lack of tumor suppressor genes mutation in neurofibromin gene on chromosome 17 (NF-1 gene) 1 in 3000 50% spontaneous mutations extremely variable expression, but 100% penetrance Major clinical features: neurofibromas - anywhere on body, in association with peripheral nerve fibers **plexiform neurofibromas pathognomonic** skin pigmentation (cafe' au lait spots) pigmented iris hamartomas (Lisch nodules) Other: skeletal changes, neurofibrosarcomas, increased risk for: Wilm's tumors, rhabdomyosarcomas, meningiomas, optic gliomas, and pheochromocytomas reduced intelligence neurofibromin downreg p21, an oncoprotein |
|
|
Neurofibromatosis Type II
|
AD, secondary to lack of tumor suppressor genes
mutations in merlin gene on chr 22 (NF-2 gene) 1 in 40,000 to 50,000 develop broad range of tumors: bilateral acoustic schwannomas multiple meningiomas gliomas, especially ependymomas Other: schwannosis, meningioangiomatosis, glial hamartomas Cafe' au lait spots maybe **BUT NO Lisch nodules** |
|
|
Mutations Image
|
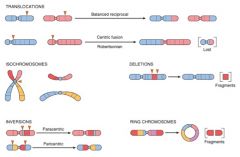
|
|
|
Cystic Fibrosis
(general) |
Cystic fibrosis (CF) is a systemic disorder of exocrine glands involving both mucus-secreting and eccrine glands throughout the body. The defects in CF involve electrolyte abnormalities and abnormally thick mucous that can cause obstruction of excretory ducts with subsequent destruction of organs, most commonly the lungs and pancreas
**The most common fatal autosomal recessive disease affecting the Caucasian population Pathophysiologic lesion is a defect in c-AMP-regulated chloride transport across apical membranes of affected epithelial cells |
|
|
CF
etiology |
Autosomal recessive inheritance
The CF gene has been localized to a single locus on the long arm of chromosome 7 The gene product is a protein called CF transmembrane conductance regulator (CFTR), which is a chloride channel that fails to be activated by c-AMP Heterozygous states have a relative resistance to diarrheal diseases, especially cholera Over 300 CF mutations that have been identified. The *most common mutation, found in approximately 70% of cases, is a deletion of a 3 base pair which causes elimination of phenylalanine in the protein product. This mutation is called delta F508 (so called because it occurs at the 508th amino acid). Those homozygous for this mutation will have the most severe form of the disease |
|
|
PKU
|
Autosomal recessive disorder
Affects people of Scandinavian descent; rarely seen in blacks or Jews Due to a severe lack of phenylalanine hydroxylase leading to hyperphenylalaninemia and PKU. Unable to convert phenylalanine to tyrosine; results in metabolites that are excreted in the urine and sweat, producing a characteristic “mousy” odor |
|
|
Maternal PKU
|
Results from the teratogenic effects of phenylalanine and/or its metabolites that cross the placenta in a female adults with PKU who don’t follow a phenylalanine restricted diet during pregnancy. Affected adults have no apparent adverse affects from elevated levels of phenylalanine and its metabolites after CNS development is complete and often abandon the low phenylalanine diet as adults. The developing fetus, whether affected by the AR defect or not, is not so fortunate, and can suffer if these dietary restrictions are not followed during pregnancy
Maternal PKU – Anamolies can include: Cardiac defects Microcephaly Low birth weight Mental retardation Slow development Language deficits |
|
|
Galactosemia
|
Autosomal recessive disorder
Deficiency in converting galactose to glucose May clinically result in hepatomegaly with jaundice, cataracts, non-specific CNS changes, failure to thrive, vomiting and diarrhea May be prevented by early removal of galactose from the diet (until at least age 2) |
|
|
X-Linked Recessive Disorders
Where on the chromosome? |
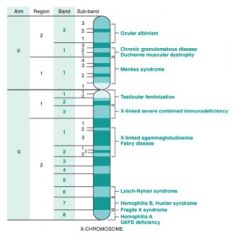
|
|
|
Multifactorial Inheritance
|
Results from the combined actions of environmental influences and the additive effects of two or more mutant genes
Risk increases with the number of abnormal genes inherited Normal phenotypic multifactoral characteristics include: Hair color, height, eye and skin color **Rate of recurrence is same for all siblings (2-7%) Includes cleft lip/palate, congenital heart disease, coronary heart disease, hypertension, gout, diabetes mellitus and pyloric stenosis |
|
|
FISH
|
Fluorescent in situ hybridization (FISH)
Can identify specific DNA sequence in interphase nuclei as well as in karyotyping studies No need for dividing cells, so can use fixed or embedded tissue *Need to know specific DNA sequences to design the probes, which are labeled with a fluorescing compound Results viewed under a fluorescent microscopic |
|
|
Trisomy 21
|
Most common of the chromosomal disorders and a major cause of mental retardation
1 in 700 live births Increased risk with increased maternal age 1 in 1550 under 20 years of age 1 in 25 over 46 years of age Most common cause is meiotic nondisjunction; 47,XX or XY, +21 (95%) Robertsonian translocation (4% of cases); 46,XX or XY, der(14;21)(q10;q10),+21 Mosaics (1% of cases)—result from mitotic nondisjunction in the zygote; 46, XX or XY/47,XX or XY,+21 Clinical findings: Flat facial profile Oblique palpebral fissures Epicanthic folds Congenital heart disease (40%) especially **endocardial cushion defects** Acute leukemia (10-20X increased risk) GI atresia, Alzheimer disease (in patients older than 40), and immunodeficiencies |
|
|
Klinefelter Syndrome
|
Male hypogonadism that occurs when there are two or more X chromosomes and at least one Y chromosome
82% are of the “classic” form; 47,XXY (90% in Robbins) 15% are mosiacs of various forms such as 46,XY/47,XXY. Have seen examples up to 49,XXXXY Slightly low IQ Atrophic testes, hypospadias, cryptorchidism and skeletal abnormalities Increased breast cancer and autoimmune disorders - Due to increased estrogen and decreased testosterone? |
|
|
XYY Males
|
47,XYY or 48,XYYY etc.
1:1000 live-born males Phenotypically normal except may be tall and have acne Normal intelligence ? Antisocial behavior Increased numbers in penal institutions |
|
|
Turner Syndrome
|
Results from complete or partial monosomy of the X chromosome
Characterized primarily by hypogonadism in phenotypic females Clinical findings: Short stature Coarctation of aorta narrowing Edema Cystic hygromas Benign tumor of dilated lymphatic channels in neck and shoulders Webbing of the neck Broad chest with widely spaced nipples Streak ovaries hypogonadism **Primary amenorrhea (causes up to 1/3 of all cases of primary amenorrhea) Infantile genitalia Hypothyroidism (50%) Insulin resistance |
|
|
Fragile X syndrome
|
Prototype of diseases in which the mutation is characterized by a long repeating sequence of 3 nucleotides - FMR-1 (familial mental retardation-1 gene)
Labeled Fragile X because the triple repeats give the X chromosome “broken” appearance on G banding 1:1550 males - The 2nd most common genetic cause of mental retardation 1:8000 females Characteristic phenotype: Long face with large mandible, large everted ears, macroorchidism (90% of postpubertal males) Less frequently seen features include: Hyperextensible joints, high arched palates and mitral valve prolapse, mimicking connective tissue disorders. “Fragile site” at Xq27.3 which upon karyotyping is seen as staining discontinuity (broken appearance) CGG is the trinucleotide repeat at this location Normal people = ~29 repeats Premutation = 50-200 repeats 30% of females have symptoms (in addition they are a carrier) Carrier males do not have any symptoms Full Mutation = 250-4000 repeats FMR-1 protein is widely expressed, but most highly in the brain and testes, explaining the syndromes most common features. Triple repeats greater than 230 lead to increased methylation of the gene which extends to the promoter region and stops production of this gene product. In the brain, FMRP complexes with mRNA to form mRNP-complexes, which are transported down the axons and regulate transcription of specific mRNA’s |
|
|
Mitochondrial Mutations
(example) |
**Leber Hereditary Optic Neuropathy
|
|
|
Genomic Imprinting
|
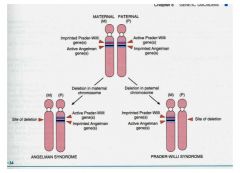
Selective inactivation of either the maternal or paternal allele, e.g. maternal imprinting refers to transcriptional silencing of the maternal allele
|