Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
206 Cards in this Set
- Front
- Back
|
What are the common features of neurodegenerative diseases? |
- Selective vulnerability of specific neurons and systems, which leads to progressive decline
- Misfolded and/or aggregated proteins - Sporadic and familial forms |
|
|
What are the characteristics of gray matter diseases?
|
Selective vulnerability of specific neurons and systems
- Progressive loss of neurons, groups of neurons, associated fiber tracts - Usually functionally related (rather than physically contagious) - Relatively symmetric |
|
|
What are some examples of neurodegenerative diseases that have selective vulnerability of specific neurons and systems? What specific neurons/systems are affected?
|
- Parkinson's Disease: Extrapyramidal system
- Alzheimer's Disease: Cerebral cortex (higher order association cortices and limbic system) - Huntington's Disease: Extrapyramidal system - Amyotrophic Lateral Sclerosis: Pyramidal system |
|
|
Which specific neurons / systems are selectively vulnerable in Parkinson's Disease?
|
Extrapyramidal system (same as Huntington's)
|
|
|
Which specific neurons / systems are selectively vulnerable in Azheimer's Disease?
|
Cerebral Cortex (higher order association cortices and limbic system)
|
|
|
Which specific neurons / systems are selectively vulnerable in Huntington's Disease?
|
Extrapyramidal system (same as Parkinson's)
|
|
|
Which specific neurons / systems are selectively vulnerable in Amyotrophic Lateral Sclerosis?
|
Pyramidal System
|
|
|
What is the common cellular hallmark of many degenerative diseases?
|
Misfolded and/or aggregated proteins
|
|
|
What are the characteristics of misfolded and/or aggregated proteins in neurodegenerative diseases?
|
- Resistant to normal degradation processes (ubiquitin-proteosome system)
- Often form inclusions (used to diagnose disease at autopsy) - Cytotoxic to neuron and/or marker of disease presence |
|
|
What are some examples of neurodegenerative diseases that have misfolded and/or aggregated proteins? What specific misfolded and/or aggregated proteins?
|
- Alzheimer's Disease: amyloid-B and tau protein
- Parkinson's Disease: a-synuclein - Frontotemporal lobar degeneration: tau, ubiquitin, TDP-43 - Huntington's Disease: polyglutamine |
|
|
What misfolded and/or aggregated proteins are present in Alzheimer's Disease?
|
- Amyloid-B
- Tau |
|
|
What misfolded and/or aggregated proteins are present in Parkinson's Disease?
|
a-Synuclein
|
|
|
What misfolded and/or aggregated proteins are present in Frontotermporal Lobar Degeneration?
|
- Tau
- Ubiquitin - TDP-43 |
|
|
What misfolded and/or aggregated proteins are present in Huntington's Disease?
|
Polyglutamine
|
|
|
In neurodegenerative diseases, are sporadic forms or familial forms more common? Exceptions?
|
- Most neurodegenerative diseases: sporadic forms more common
- Huntington's disease: only autosomal dominant form |
|
|
What is the benefit of genetic linkage studies in familial forms of neurodegenerative disease?
|
- Makes candidate genes and proteins easier to identify
- Aids in deciphering mechanisms (often applicable to both sporadic and familial disease types) |
|
|
What are the possible etiologies of neurodegenerative diseases?
|
- Genetic mutations
- Genetic polymorphisms (risk factors) - Aging - Environmental toxins |
|
|
What are the cellular mechanisms that contribute to development of neurodegenerative diseases?
|
- Oxidative stress and generation of ROS
- Inflammation - Disruption of axonal transport and synaptic function - Dysfunctional waste clearance (inhibition of ubiquitin proteosome system and autophagy) - Mitochondrial dysfunction - Programmed cell death (apoptosis) |
|
|
What does the term "selective vulnerability" mean in relation to neurodegeneration?
|
Some neurons are more vulnerable to toxic insults than others; this is true even when the gene responsible is widespread (eg, Huntington's disease)
|
|
|
What types of insults contribute to induction of neurodegeneration?
|
- Environmental toxins
- Neuronal metabolism (oxidative) - Aging |
|
|
How do environmental toxins induce neurodegeneration?
|
- Reduce mitochondrial function
- Decrease ATP production - Increase oxidative stress |
|
|
How does neuronal metabolism contribute to neurodegeneration?
|
Neuronal metabolism is oxidative
|
|
|
How does aging contribute to neurodegeneration?
|
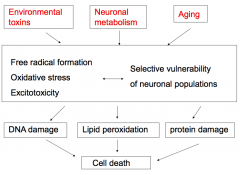
- Affects mitochondrial function
- Loss of protective enzymes and molecules - Progressive hits accumulate with aging |
|
|
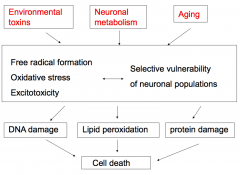
What are the collective implications of environmental toxins, neuronal metabolism, and aging, as it relates to neurodegeneration?
|
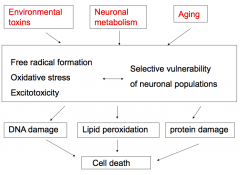
- Formation of free radicals
- Oxidative stress - Excitotoxicity - Selective vulnerability of neuronal populations These lead to: - DNA damage - Lipid peroxidation - Protein damage Ultimately: - Cell death |
|
|
What leads to free radical formation? How can that cause neurodegeneration?
|
Dysfunctional mitochondria:
- Toxins and aging lead to loss of mitochondrial function - Inefficient mitochondrial electron transport leads to leaking of electrons and oxygen radicals - Complex 1 is vulnerable to injury in response to free radicals - Free radicals cause lipid peroxidation and loss of membrane integrity |
|
|
Which part of the mitochondria is particularly vulnerable to injury by free radicals?
|
Complex 1 is vulnerable to injury in response to free radicals
|
|
|
Which molecules cause oxidative stress that contributes to neurodegeneration? How are these molecules formed?
|
- Hydrogen Peroxide and Superoxide
- Oxygen is easily converted to these reactive molecules by the same enzymes that utilize oxygen as fuel (leaky and inefficient) - Precipitating factor for both excitotoxicity and mitochondrial dysfunction; also results from both excitotoxicity and mitochondrial dysfunction |
|
|
What causes excitotoxicity?
|
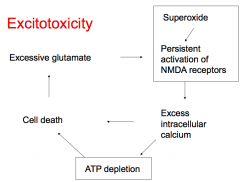
- Superoxide leads to persistent activation of NMDA receptors
- This leads to excess intracellular Ca2+, ATP depletion, and cell death - Cell death leads to excessive glutamate which also causes persistent activation of NMDA receptors |
|
|
What leads to persistent activation of NMDA receptors?
|
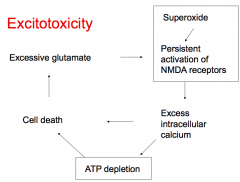
- Superoxide
- Excessive glutamate |
|
|
What does persistent activation of NMDA receptors cause?
|
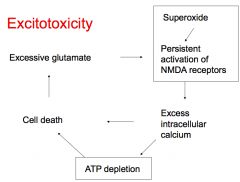
- Excess intracellular Ca2+
- ATP depletion - Cell death - Excessive glutamate (which continues to stimulate NMDA receptors) |
|
|
What are the mechanisms cells have to deal with oxyradicals? Why do these mechanisms not work in neurodegenerative disease?
|
Mechanisms to deal with oxyradicals:
- Ascorbate - Glutathione - Superoxide dismutase - Catalase (inactivates hydrogen peroxide) Degeneration occurs when the cells either produce too many radicals or lose the ability to detoxify them |
|
|
What is the most common form of dementia?
|
Alzheimer's Disease (5.2 million people in US in 2013)
|
|
|
What influences the incidence of Alzheimer's Disease?
|
- Age related: doubles every 5y after age 65
- Prevalence increases with age and affects up to 50% of people >85 years |
|
|
How long does Alzheimer's Disease last?
|
Varies from 2 to 20 years
|
|
|
What is the clinical definition of Alzheimer's Disease?
|
- Gradual and progressive decline in cognitive function
- Impairment in recent memory and one additional cognitive domain that is not d/t other medical or psychiatric illness - Results in functional impairment socially or occupationally |
|
|
What cognitive domains can Alzheimer's Disease affect?
|
- Memory
- Language - Abstract thinking and judgment - Visuo-spatial or perceptual skills - Praxis - Executive function |
|
|
What is DAT?
|
Dementia of Alzheimer type
|
|
|
What are the diagnostic criteria for Definite DAT (Dementia of Alzheimer type)? Accuracy?
|
- Clinical criteria for Probable DAT
- Histopathologic evidence of DAT (autopsy or biopsy) Accurate clinical dx in 80-90% |
|
|
What are the diagnostic criteria for Probable DAT (Dementia of Alzheimer type)?
|
- Dementia
- Two areas of cognitive impairment - Progression over time - Normal sensorium - Age of onset between 40-90 years - No other disease contributing to the dementia |
|
|
When does Probable DAT (Dementia of Alzheimer type) begin?
|
Between 40-90 years
|
|
|
What are the diagnostic criteria for Possible DAT (Dementia of Alzheimer type)?
|
- Atypical onset, presentation, or progression of dementia syndrome without known etiology
- Systemic or other brain disease capable of producing dementia - Gradually progressive decline in a single intellectual function in the absence of any other identifiable cause |
|
|
What are the principal clinical findings in Stage 1 of DAT (Dementia of Alzheimer type)? Duration?
|
- Memory: new learning is defective, remote recall is mildly impaired
- Visuospatial skills: topographic disorientation, poor complex construction - Language: poor wordlist generation, anomia - Psychiatric features: depression, delusions * Duration: 1-3 years |
|
|
What are the principal clinical findings in Stage 2 of DAT (Dementia of Alzheimer type)? Duration?
|
- Memory: recent and remote recall more severely impaired
- Visuospatial skills: poor construction, spatial disorientation - Calculation: acalculia - Psychiatric features: delusions * Duration: 2-10 years |
|
|
What are the principal clinical findings in Stage 3 of DAT (Dementia of Alzheimer type)? Duration?
|
- Intellectual functions: severely impaired
- Sphincter control: urinary and fecal incontinence - Motor: limb rigidity and flexion posture * Duration: 8-12 years |
|
|
What is the gross appearance of the brain in Alzheimer's Disease?
|
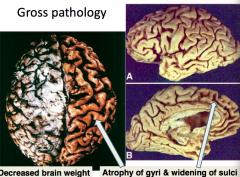
- Decreased brain weight
- Atrophy of gyri - Widening of sulci - Increased size of lateral ventricles |
|
|
What can the increased size of the lateral ventricles in Alzheimer's Disease lead to?
|
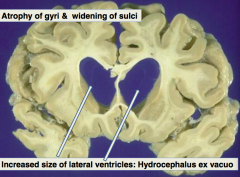
Hydrocephalus ex Vacuo
|
|
|
What is the histologic appearance of the brain in Alzheimer's Disease?
|
Senile plaques:
- Diffuse plaques - Neuritic plaques - Cerebral amyloid angiopathy - Neurofibrillary tangles (NFT) |
|
|
What causes Diffuse Plaques in Alzheimer's Disease?
|
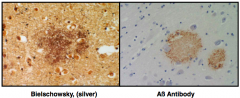
- Extracellular accumulation of Aβ protein
- Comes from Amyloid Precursor Protein (APP) |
|
|
What causes Neuritic Plaques in Alzheimer's Disease?
|
- Extracellular accumulation of Aβ protein and tau containing neurites
- Neurites are axons or dendrites coursing through brain tissue |
|
|
How do Diffuse Plaques and Neuritic Plaques compare?
|
Diffuse Plaque:
- Extracellular accumulation of Aβ protein Neuritic Plaque: - Extracellular accumulation of Aβ protein and tau containing neurites - More closely associated with cognitive decline than the diffuse plaque |
|
|
Where are senile plaques (diffuse and neuritic) more commonly found?
|
- Especially in temporal lobe (amygdala and hippocampus)
- Neocortex: senile plaques |
|
|
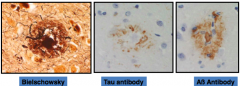
What microscopic finding is almost always found in Alzheimer's Disease, but may also occur in the absence of AD?
|
Cerebral Amyloid Angiopathy
|
|
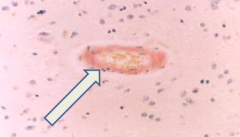
What is Cerebral Amyloid Angiopathy associated with?
|
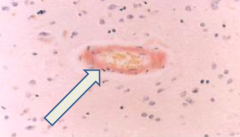
- Almost always found in AD
- Occurs in absence of AD too (associated with lobar hemorrhages in elderly) |
|
|
What causes neurofibrillary tangles (NFT)?
|
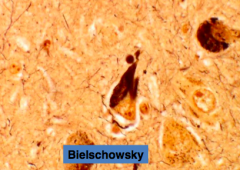
Intraneuronal accumulation of abnormally phosphorylated form of tau, a normal microtubule associated protein
|
|
|
Where are neurofibrillary tangles found?
|
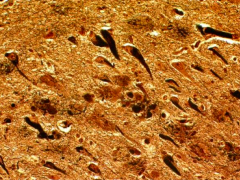
Hippocampus
|
|
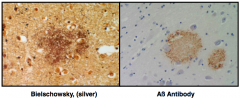
What is this?
|
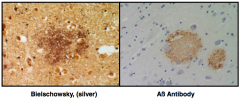
Diffuse Plaque
– Extracellular accumulation of Aß protein – Normal protein whose function is not yet completely known – Comes from Amyloid precursor protein |
|
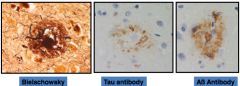
What is this?
|
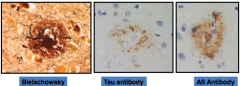
Neuritic Plaque
- Extracellular accumulation of Aß protein and tau containing neurites - More closely associated with cognitive decline than the diffuse plaque - Neurites are axons or dendrites coursing through brain tissue |
|
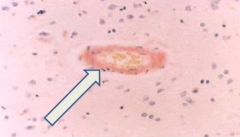
What is this?
|
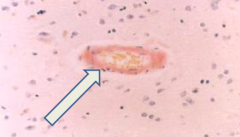
Cerebral Amyloid Angiopathy
• Almost always found in AD • Occurs in absence of AD also – Associated with lobar hemorrhages in elderly |
|
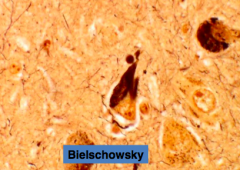
What is this?
|
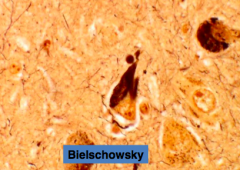
Neurofibrillary Tangles (NFT)
• Intraneuronal accumulation of an abnormally phosphorylated form of tau, a normal microtubule associated protein • NFT are not unique to AD; also found in other degenerative diseases (will discuss later in lecture) • No evidence of mutations of tau gene |
|
|
How do you make the pathological diagnosis of Alzheimer's Disease?
|
- Density of neuritic plaques
- Staging scheme for neurofibrillary tangles (NFT) |
|
|
What is the biggest risk factor for Alzheimer's Disease?
|
Age
|
|
|
What are the patterns of inheritance of Alzheimer's Disease?
|
- 75% sporadic
- 20-25% have history of affected relatives who develop disease randomly - 1-5% prominent family history, usually consistent with autosomal dominant inheritance pattern (FAD = familial AD) |
|
|
How often is Alzheimer's Disease occurring sporadically?
|
75%
|
|
|
How often is Alzheimer's Disease occurring to patients with a history of affected relatives who develop disease randomly?
|
20-25%
|
|
|
How often is Alzheimer's Disease occurring to patients with a prominent family history, usually consistent with an autosomal dominant inheritance (familial Alzheimer's Disease = FAD)?
|
1-5%
|
|
|
What are the characteristics of EARLY-onset Alzheimer's Disease?
|
Different from late-onset:
* <60-65 years * Often autosomal dominant mutation, highly penetrant Same as late-onset: - Memory impairment followed by cognitive decline - Neuritic plaques and neurofibrillary tangles |
|
|
What are the characteristics of LATE-onset Alzheimer's Disease?
|
Different from early-onset:
* >60-65 years * No Mendelian pattern of inheritance Same as early-onset: - Memory impairment followed by cognitive decline - Neuritic plaques and neurofibrillary tangles |
|
|
Which protein is the precursor for β amyloid protein, Aβ, (in senile plaques)?
|
Amyloid Precursor Protein
|
|
|
What kind of protein is Amyloid Precursor Protein (APP)? What chromosome is it on? Significance?
|
- Transmembrane glycoprotein
- Gene on chromosome 21 - β amyloid protein is derived from APP |
|
|
What is the link between Alzheimer's Disease and Down's Syndrome?
|
- Individuals with Down's Syndrome (Trisomy 21) develop AD in late 30s
- Amyloid Precursor Protein (APP) is found on chromosome 21, so having a trisomy of 21 increases the amount of β amyloid protein |
|
|
How is Amyloid Precursor Protein processed?
|
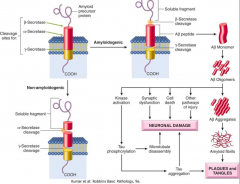
- α-Secretase cleaves within the Aβ sequence, so no Aβ is produced
- Cleavage at β and γ sites of APP: Aβ produced, β secretase enzyme identified, γ secretase enzyme is unknown (presenilin proteins may be cofactors or enzymes) |
|
|
What enzyme prevents Aβ (β Amyloid Protein) from being formed?
|
α Secretase - cleaves within Aβ sequence
|
|
|
How does Aβ (β Amyloid Protein) get produced?
|
Cleavage at β and γ sites of APP:
- Aβ produced - β Secretase enzyme identified - γ Secretase enzyme unknown |
|
|
What is the most commonly occurring genetic mutation in Alzheimer's Disease? Chromosome?
|
Presenilin 1 (PSEN1 gene) found on chromosome 14
|
|
|
What kind of protein is Presenilin 1? Chromosome? Importance?
|
- Transmembrane protein (ER and Golgi)
- Found on chromosome 14 - Most commonly occurring genetic mutation in AD |
|
|
Mutations in what genes are important for Alzheimer's Disease? How are these mutations inherited? Results?
|
Genes:
- Presenilin 1 (PSEN1) - most commonly occurring genetic mutation - Amyloid Precursor Protein (APP) - rare - Presenilin 2 (PSEN2) - very rare Inheritance: autosomal dominant Results in increased deposition of Aβ amyloid protein, which leads to early onset AD (<65 years) |
|
|
What is the only established genetic RISK FACTOR for late-onset Alzheimer's Disease? Effect?
|
Apolipoprotein E protein (APOE gene) - increases Aβ deposition
|
|
|
What alleles on Apolipoprotein E protein (APOE gene) affect the risk for late-onset Alzheimer's Disease? Chromosome?
|
Three alleles at gene locus on chromosome 19: ε2, ε3, and ε4
- Codes three protein isoforms: E2, E3, and E4 - No associated mutations in gene |
|
|
What is the function of the Apo# protein?
|
Cholesterol transport, metabolism, and storage
|
|
|
How does the presence of the ε4 allele modify the genetic risk for Alzheimer's Disease?
|
- Decreases age on onset of AD
- Dose-dependent - Association robust but not specific - Presence of ε4 not necessary nor sufficient |
|
|
What diseases is a cholinergic signaling deficiency involved in?
|
- Alzheimer's Disease
- Dementia with Lewy Bodies - Vascular Dementia |
|
|
What is the relationship between the treatment of Alzheimer's Disease, Dementia with Lewy Bodies, and Vascular Dementia?
|
Cholinergic Signaling Deficiency
|
|
|
What drugs can be used to modify the Cholinergic Signaling deficiency in Alzheimer's Disease, Dementia with Lewy Bodies, and Vascular Dementia?
|
Centrally-acting cholinesterase inhibitors:
- Donepezil - Rivastigmine - Galantamine |
|
|
What are the centrally-acting cholinesterase inhibitors?
|
- Alzheimer's Disease
- Dementia with Lewy Bodies - Vascular Dementia |
|
|
What is the mechanism and efficacy of Donepezil, Rivastigmine, and Galantamine?
|
- Centrally-acting cholinesterase inhibitors
- Modest improvement in Alzheimer's Disease, Dementia with Lewy Bodies, and Vascular Dementia |
|
|
What drug can modify the excitotoxicity, oxidative stress, and neuroinflammation in Alzheimer's Disease?
|
Memantine
|
|
|
What is the mechanism and efficacy of Memantine?
|
- Mechanism: NMDA channel blocker
- Some efficacy in slowing disease progression of AD |
|
|
What is the second most common form of early dementia (<65 years) after Alzheimer's Disease?
|
Frontotemporal Degeneration (FTD)
|
|
|
What does Frontotemporal Degeneration (FTD) cause?
|
- Focal degeneration in the frontal and anterior temporal lobes
- Second most common form of early dementia (onset <65 years) |
|
|
What is the term for the pathologic entity of Frontotemporal Degeneration (FTD)?
|
Frontotemporal Lobar Degeneration (FTLD)
|
|
|
How common are sporadic and familial cases of Frontotemporal Degeneration (FTD)? Inheritance?
|
Sporadic: ~50%
Familial: autosomal dominant |
|
|
What are the clinical subtypes of Frontotemporal Degeneration (FTD)? How common?
|
Behavioral variant FTD (vbFTD): 50%
Primary progressive aphasia: - Progressive non-fluent aphasia (PNFA): 25% - Semantic variant (SV): 20-25% |
|
|
What gross findings are associated with the behavioral variant of Frontotemporal Degeneration (bvFTD)? How common is this subtype?
|
- Bi-frontal lobe atrophy
- 50% of cases |
|
|
What gross findings are associated with the Progressive Non-Fluent Aphasia subtype (PNFA) of Frontotemporal Degeneration (FTD)? How common is this subtype?
|
- Left peri-Sylvian atrophy
- 25% of cases |
|
|
What gross findings are associated with the Semantic Variant (SV) subtype of Frontotemporal Degeneration (FTD)? How common is this subtype?
|
- Bilateral anterior temporal lobe atrophy
- 20-25% of cases |
|
|
What are the symptoms of the Behavioral Variant Frontotemporal Degeneration (bvFTD)?
|
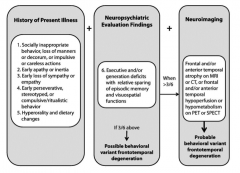
- Socially inappropriate behavior, loss of manners or impulsive or careless actions
- Early apathy or inertia - Early loss of sympathy or empathy - Early perseverative, stereotyped, or compulsive/ritualistic behavior - Hyperorality and dietary changes |
|
|
What are the neuropsychiatric evaluation findings of the Behavioral Variant Frontotemporal Degeneration (bvFTD)?
|
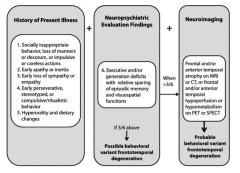
Executive and/or generation deficits with relative sparing of episodic memory and visuospatial functions
|
|
|
When would you do neuroimaging in a patient you suspect has Behavioral Variant Frontotemporal Degeneration (bvFTD)? What are the neuroimaging signs that would support your diagnosis?
|
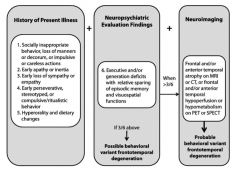
- >3/6 of the history of present illness and neuropsychiatric evaluation findings
Neuroimaging findings: - Frontal and/or anterior temporal atrophy on MRI or CT - Frontal and/or anterior temporal hyperperfusion or hypometabolism on PET or SPECT |
|
|
What are the primary progressive aphasia subtypes of FrontoTemporal Degeneration (FTD)?
|
- Fluency: worst in non-fluent aphasia
- Comprehension: worst in semantic variant - Repetition |
|
|
What are the gross findings of Frontotemporal Lobar Degeneration (FTLD)?
|
Frontal and temporal lobe atrophy
|
|
|
What are the microscopic findings of Frontotemporal Lobar Degeneration (FTLD)?
|
- Tauopathies: accumulation of tau protein
- FTLD-TDP-43: accumulation of TDP-43 |
|
|
What is the classic example of accumulation of tau protein? Gross and microscopic findings?
|
Pick's Disease
- Atrophy of frontal and temporal lobes - Severe neuronal loss in frontal and temporal cortices - Pick bodies: round cytoplasmic inclusion in neurons, containing abnormal au filaments |
|
|
What are Pick Bodies?
|
Round cytoplasmic inclusion in neurons that contain abnormal tau filaments
|
|
|
Where are there accumulations of TDP-43 in Frontotemporal Lobar Degeneration (FTLD)?
|
Cytoplasmic protein accumulations in frontal and temporal lobes
|
|
|
What is the gross appearance in Pick's Disease?
|
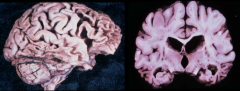
Distinctive atrophy: severe frontal and temporal atrophy ("Knife-edge"), sparing of parietal and occipital lobes
|
|
|
What is the microscopic appearance in Pick's Disease?
|
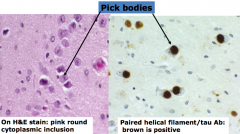
Pick Bodies: round cytoplasmic inclusion in neurons, contain abnormal tau filaments
|
|
|
What are the gross and microscopic findings in Frontotemporal Lobar Degeneration (TDP-43)?
|
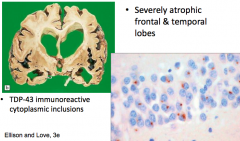
- Severely atrophic frontal and temporal lobes
- TDP-43 immunoreactive cytoplasmic inclusions |
|
|
What is the clinical presentation of Parkinsonism?
|
- Rigidity
- Bradykinesia (slowed movements) - Tremor (usually resting) - Mask facies - Stooped posture - Festinating gait (progressively shortened accelerated steps) |
|
|
What is the clinical differential diagnosis for these symptoms:
- Rigidity - Bradykinesia (slowed movements) - Tremor (usually resting) - Mask facies - Stooped posture - Festinating gait (progressively shortened accelerated steps) |
- Parkinson's Disease (idiopathic) - most common cause
- Multiple System Atrophy - Progressive Supranuclear Palsy - Corticobasal Degeneration - Intoxication with MPTP - Post-encephalitic Parkinson's Disease |
|
|
How common is Parkinson's Disease? Who is more likely to get it?
|
- 500,000 to 1,000,000 patients in US
- 1-in-40 lifetime risk - Slight male predominance - Prevalence increases with age (peak onset: 55-65 years) - Positive family history in 6-16% of PD cases |
|
|
What is the strongest risk factor for Parkinson's Disease?
|
Age: peak onset is 55-65 years
|
|
|
How often is their a family history for Parkinson's Disease?
|
6-16% of PD cases:
- If have 1 first degree relative w/ PD, 3x greater risk - If have 2+ first degree relatives w/ PD, 10x greater risk |
|
|
What are the possible pathogenetic mechanism to explain Parkinson's Disease?
|
No unifying mechanism yet, possibilities:
- Misfolded protein / stress response triggered by protein aggregation - Defective proteosomal function - Altered mitochondrial function |
|
|
What is the genetic relationship to Parkinson's Disease?
|
- Predominantly sporadic
- Familial cases: mutations in 5 genes (Parkin and α-Synuclein gene) |
|
|
Genes involved in familial cases of Parkinson's Disease have what functions?
|
- Synaptic function
- Protein degradation - Mitochondrial function |
|
|
What chromosome is the Parkin gene found on? Significance?
|
- Chromosome 6
- Younger onset Parkinson's Disease - 6% of all families screened had mutations |
|
|
What is the significance of the α-Synuclein gene in Parkinson's Disease?
|
- Very rare mutation present in families with autosomal dominant familial Parkinson's Disease
- Subsequently discovered to be a major component of Lewy Bodies - Sporadic PD: no mutation in α-Synuclein gene - Linkage between α-Synuclein and disease pathogenesis is not understood |
|
|
What environmental toxins are related to Parkinson's Disease?
|
- Manganese
- Carbon monoxide - Rural living: well-water drinking - Paper mills - Hydrocarbons (petroleums, plastics) - Residential use of pesticides - Protective (?): cigarette smoking and caffeine |
|
|
What are the clinical features of Parkinson's Disease?
|
TRAP:
- Tremor (resting) - Rigidity - Akinesia (or bradykinesia - slowness) - Postural changes (gait/balance problems) |
|
|
What is the most common presenting symptom of Parkinson's Disease? Characteristics?
|
Tremor:
- Typically asymmetric and low in frequency (4Hz to 6Hz) - Pill-rolling type of movements - Most often affects the distal upper extremities - Chin tremor specific for PD |
|
|
What are the characteristics of bradykinesia in Parkinson's Disease?
|
- Slowness or lack of movement
- Micrographia: abnormally small, cramped handwriting - Hypophonia: soft speech - Hypomimia: reduced facial expression - Decreased blink rate - Decreased arm swing with walking - Shortened stride length |
|
|
What are the characteristics of rigidity in Parkinson's Disease?
|
Increased resistance to passive movement of skeletal muscle across a joint
|
|
|
What are the characteristics of postural instability and gait disturbance in Parkinson's Disease?
|
- Early: manifests as dragging of one leg or stooped posture
- Late: gait problems can be severe |
|
|
What are the non-motor symptoms in the pre-motor phase of Parkinson's Disease?
|
- Sleep: REM behavior disorder
- Olfactory loss - Dysautonomia |
|
|
What are the supportive criteria for a diagnosis of Parkinson's Disease?
|
- Unilateral onset
- Rest tremor present - Excellent response to levodopa - Clinical course for >10 years |
|
|
What are the confirmatory tests for a diagnosis of Parkinson's Disease?
|
- No lab tests
- No blood tests - No x-rays or MRI scans * Must be a response to L-DOPA |
|
|
What are the gross and microscopic findings of Parkinson's Disease?
|
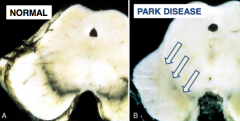
- Pallor of substantia nigra
- Neuronal loss and Lewy bodies in substantia nigra - Lewy bodies: eosinophilic cytoplasmic neuronal inclusions that contain α-Synuclein |
|
|
What are the diagnostic characteristics of Dementia with Lewy Body (DLB)?
|
Dementia associated with any two of the following core clinical features:
- Fluctuating cognition or level of consciousness - Visual hallucinations - Parkinsonian motor signs |
|
|
What are the suggestive characteristics of Dementia with Lewy Body (DLB)?
|
- Rapid Eye Movement (REM) sleep behavior disorder
- Neuroleptic sensitivity - Low dopamine transporter uptake in the basal ganglia (If one or more of these suggestive features is present along with one or more core features, a diagnosis of PROBABLE dementia with Lewy Body (DLB) can be made) |
|
|
What are the similarities and differences of Dementia with Lewy Body (DLB) and Parkinson's Disease?
|
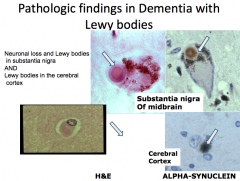
Both:
- Substantia nigra degeneration and Lewy bodies DLB: - Clinical features of dementia PD: - Parkinsonian signs and symptoms - Most eventually develop dementia (5x more frequent than age-matched controls) - Cortical Lewy Bodies |
|
|
What are the goals of Parkinson's Disease pharmacotherapy?
|
Primarily targeted to restoring the neurotransmission in the circuit that regulates the extrapyramidal system
|
|
|
What is the etiology of Parkinson's Disease?
|
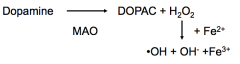
- Dopaminergic cells of the substantia nigra are selectively vulnerable
- Dopamine when metabolized leads to the production of free radicals - As cells age, the accumulation of years of free radicals reduces the number of dopamine cells |
|
|
What is the life cycle of Dopamine?
|
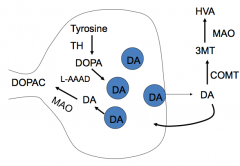
Synthesis:
- Tyrosine → DOPA via Tyrosine Hydroxylase - DOPA → Dopamine via L-AAAD (decarboxylase) Breakdown: - DA → DOPAC via MAO - DA → 3MT via COMT; 3MT → HVA via MAO |
|
|
What is the mechanism of Levodopa (L-DOPA)? Rationale for use?
|
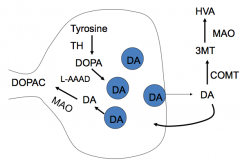
- Mechanism: uses amino acid transporters to enter the brain; decarboxylated to DA in DA cells and other neurons that express L-AAAD (L-aromatic amino acid decarboxylase)
- Rationale: restores levels of DA in the basal ganglia |
|
|
What are the pharmacokinetic considerations of Levodopa (L-DOPA)? Considerations?
|
- At most, 2% of an oral dose of L-DOPA gets into the brain
- Most L-DOPA is metabolized to DA by peripheral L-AAAD (in liver) or COMT (in gut) * Lots of dopamine where you don't want it, leading to side effects * Not enough gets into the brain - Half-life of L-DOPA is very short (1-3 hours) - Absorption is dependent on GI contents, it is taken up by amino acid transporters that can be saturated after a high protein meal |
|
|
What causes only 2% of Levodopa (L-DOPA) from entering the brain? Implications?
|
Most L-DOPA is metabolized to DA by peripheral:
- L-AAAD in the liver - COMT in the gut * Lots of dopamine where you don't want it, leading to side effects |
|
|
How do you prevent Levodopa (L-DOPA) from being metabolized in the periphery by L-AAAD in the liver and COMT in the gut?
|
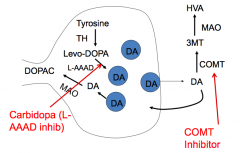
Co-administer with Carbidopa
- Carbidopa inhibits L-AAAD, but does not cross the blood brain barrier Co-administer with Entacapone - Entacapone is a COMT inhibitor |
|
|
What is the mechanism and utility of Carbidopa?
|
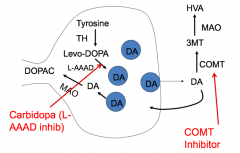
- Mechanism: inhibits L-AAAD in the periphery
- Utility: increases the dose of L-DOPA that gets to CNS, decreasing peripheral side effects |
|
|
What is the mechanism and utility of Entacapone?
|
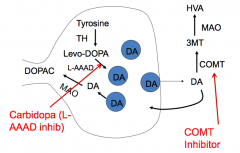
- Mechanism: COMT inhibitor
- Utility: increases DA concentration in brain |
|
|
How can the meal you eat affect the absorption of L-DOPA?
|
After a high protein meal, the amino acid transporters that are responsible for uptake of L-DOPA may be saturated
|
|
|
What is the clinical usefulness of L-DOPA?
|
- Very effective, if it doesn't help, it probably isn't PD
- Effective against all of the symptoms, especially bradykinesia - Early disease, beneficial effects outlast half-life which suggests that the DA vesicles are filled; effects are sustained and smooth * Best results obtained in first few years of treatment (later, efficacy is lost or becomes erratic regardless of disease intensity) |
|
|
How long is Levodopa (L-DOPA) effective? Implications for its use?
|
- Best results are obtained in the first few years (therefore, DO NOT USE until the symptoms cause functional impairment)
- After a few years, the efficacy is lost or becomes erratic (regardless of disease intensity) |
|
|
What are the CNS side effects of Levodopa (L-DOPA)? Why do they occur? Implications?
|
Due to conversion to DA
- Wearing off d/t L-DOPA loss from body - Dyskinesias: too much movement - Dementia, confusion: treat w/ atypical antipsychotics (clozapine and quetiapine) * CNS effects not alleviated by co-administration w/ Carbidopa |
|
|
How do you treat dementia and confusion in patients with Parkinson's Disease being treated with L-DOPA?
|
Atypical anti-psychotics (Clozapine and Quetiapine)
|
|
|
What are the PNS side effects of Levodopa (L-DOPA)?
|
Due to conversion to DA
- GI: nausea - CV: postural hypotension (common), arrhythmias (rare), and hypertension * CNS effects greatly reduced by co-administration w/ Carbidopa |
|
|
Which drugs does L-DOPA interact with?
|
- Pyridoxine (vitamin B6)
- MAO inhibitors - Halothane - Anti-psychotics that are DA receptor antagonists |
|
|
Why shouldn't patients on L-DOPA be taking Vitamin B6?
|
Vitamin B6 increases metabolism of DA in the periphery
|
|
|
Why shouldn't patients on L-DOPA be taking MAO inhibitors?
|
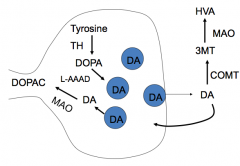
Can cause hypertension (exception: selegiline)
|
|
|
Why shouldn't patients on L-DOPA be taking Halothane?
|
Sensitizes the heart to arrhythmias
|
|
|
What are the contraindications to using L-DOPA?
|
- Glaucoma
- Psychosis - Cardiac disease that involves arrhythmias - Malignant melanoma |
|
|
What are the Dopamine Receptor Agonists?
|
- Pramipexole and Ropinerole (selective D2 agnoists)
- Apomorphine (high affinity D4 agonist, moderate affinity for D2, D3, and D5) |
|
|
What is the mechanism and rationale for using Dopamine Receptor Agonists?
|
- Rationale: mimics dopamine without the need for intact nerve terminals
- Mechanism: agonists of DA receptors in the striatum |
|
|
What are the advantages of Dopamine Receptor Agonists over L-DOPA?
|
- No enzymatic conversion is needed
- Selectively for receptor subtypes - Longer half-life - Less DA-dependent oxidative stress (?) |
|
|
What is the mechanism of Pramipexole and Ropinerole?
|
- Selective D2 agonists
- Can be titrated to therapeutic doses over a week or less |
|
|
What is the mechanism of Apomorphine?
|
- High affinity for D4 agonist
- Moderate affinity for D2, D3, and D5 - Used by subcutaneous injection for immediate therapy of an "off" episode (Reserved for patients refractory to other treatments) |
|
|
What are the first line treatments for Parkinson's Disease in young patients? Elderly?
|
Young patients: direct dopamine agonists
- Pramipexole and Ropinerole (selective D2) - Apomorphine (high affinity for D4, moderate affinity for D2, D3, and D5) Elderly patients: L-DOPA (avoid direct dopamine agonists because the confusion is worse) |
|
|
What are the benefits of Direct Dopamine Agonists over L-DOPA for the first-line treatment of Parkinson's Disease?
|
- Longer duration of action (half-life) than L-DOPA results in fewer end-of-dose problems, less hyperactivity
- Reduced DA synthesis, less oxidative toxicity |
|
|
What drug is reserved for patients with Parkinson's Disease that are refractory to other treatments?
|
Apomorphine (high affinity D4 agonist, moderate affinity for D2, D3, and D5)
|
|
|
What are the side effects of direct dopamine agonists?
|
- Nausea (especially bromocriptine)
- Fatigue - Sudden attacks of daytimes sleep - CNS toxicity: confusion (worse than L-DOPA) and dyskinesia (not as bad as L-DOPA) - Apomorphine can increase QT prolongation and can cause injection site reactions |
|
|
How do MAO inhibitors affect Parkinson's Disease?
|
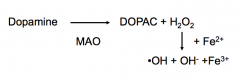
MAO Inhibitor will increase the amount of DA and decrease oxidative stress on the neuron (potentially protective?)
|
|
|
Why are MAO inhibitors for Parkinson's Disease a problematic therapy?
|
- MAO in the liver is essential for metabolizing tyramine that is ingested
- Tyramine is a sympathomimetic |
|
|
How can you avoid the sympathomimetic affects of tyramine with MAO inhibitors?
|
Use MAO-B inhibitors
- MAO-A is the major form found in the human GI tract and liver (where tyramine is metabolized) - MAO-B is the major form found in the brain |
|
|
Which form of MAO should be selectively inhibited to treat Parkinson's Disease?
|
- MAO-B because this is the major form found in the brain
- MAO inhibition will prolong the action of dopamine and may reduce oxidative stress on neurons |
|
|
What drugs are selective, irreversible inhibitors of MAO-B?
|
- Selegiline
- Rasagiline |
|
|
What are the clinical uses of Selegiline and Rasigiline (MAO-B inhibitors)?
|
- Usually prescribed as as soon as the disease is diagnosed, with modest benefit
- In more advanced disease, used w/ L-DOPA to prolong its effective half-life - Anti-depressent (increases NE) - Modest effects on disease progression |
|
|
What are the side effects of Selegiline and Rasigiline (MAO-B inhibitors)?
|
- Generally well tolerated in early disease
- In advanced disease, worsens the side effects of L-DOPA (too much DA?) - Metabolized to amphetamine and methamphetamine which may cause anxiety and insomnia; reduced when administered as an orally disintegrating tablet or as a patch (avoid first pass metabolism) |
|
|
Which drugs can cause anxiety and insomnia d/t metabolism to amphetamine and methamphetamine? How can this be avoided?
|
- Selegiline and Rasagiline
- Reduce the anxiety and insomnia when administered as an orally disintegrating tablet or as a patch (these avoid the first-pass metabolism) |
|
|
Selegiline and Rasagiline (MAO-B inhibitors) can cause adverse drug interactions when administered with what other drugs? What can this lead to?
|
- Meperidine
- Tricyclic anti-depressants - Serotonin Reuptake Inhibitors - Can lead to Serotonin Syndrome (stupor, rigidity, agitation, and hyperthermia) |
|
|
What are the COMT inhibitors?
|
- Tolcapone
- Entacapone |
|
|
What is the mechanism and rational for Tolcapone and Entacapone?
|
- COMT inhibitors
- COMT metabolizes dopamine so its inhibition will prolong the action of DA, it also increases L-DOPA metabolism to non-DA metabolites |
|
|
What are the similarities and differences of Tolcapone and Entacapone?
|
Both:
- COMT inhibitors Tolcapone: - Hepatotoxicity limits utility - Long half-life - Inhibits both peripheral and central COMT (all the benefits) Entacapone: - Short half-life - Does not get into CNS well so mainly inhibits peripheral COMT - Co-administer with L-DOPA to eliminate peripheral metabolism (like Carbidopa) |
|
|
What are the side effects of Tolcapone and Entacapone (COMT inhibitors)?
|
- Common (d/t decreased dopamine: nausea, orthostatic hypotension, vivid dreams, confusion, and hallucinations
- Tolcapone can produce liver toxicity, not used except as a last resort and with hepatic monitoring!! |
|
|
What are the anti-muscarinic drugs used for Parkinson's Disease?
|
- Trihexyphenidyl
- Benztropine |
|
|
What is the mechanism and rationale for using Trihexyphenidyl and Benztropine?
|
- Anti-muscarinics: antagonize striatal muscarinic receptors
- Cholinergic interneurons in the striatum are normally inhibited by DA, the loss of DA results in overactivity of these excitatory neurons (blunted by anti-cholinergics) |
|
|
What is the effectiveness and clinical usefulness of Trihexyphenidyl and Benztropine (anti-muscarinics)?
|
- Modest efficacy: not good against bradykinesia
- 3rd choice (after L-DOPA and DA agonists), used when dopaminergic therapy is contraindicated, early disease, elderly - Can be used in combo w/ L-DOPA/DA agonists so that those doses can be lower |
|
|
What are the side effects of Trihexyphenidyl and Benztropine (anti-muscarinics)?
|
- Sedation, mental confusion
- Atropine-like side effects in periphery |
|
|
What is the mechanism and rational of Amantadine?
|
- Increases dopamine release, mildly anti-cholinergic; blocks NMDA receptors
- Originally used as an anti-viral agent |
|
|
How effective is Amantadine for Parkinson's Disease? Which other drugs can it be given with, why?
|
- Less effective w/ short-lived benefits
- Often given w/ L-DOPA (esp if there are end-of-dose problems) or with anti-cholinergics - Not useful if L-DOPA is ineffective |
|
|
What are the side effects of Amantadine? Contraindications?
|
- Dizziness, lethargy, sleep disturbances
- Can cause peripheral edema - Sympathomimetic effects d/t effects on catecholamine release - Contraindicated in patients with congestive heart failure |
|
|
How is Huntington Disease inherited? Mutation?
|
Autosomal Dominant
- Mutation on huntingtin gene on chromosome 4 - Expanded trinucleotide repeat in gene (CAG) causes structural abnormalities |
|
|
What is the presentation of Huntington Disease at onset?
|
- Motor symptoms are predominant in 60% of cases
- Chorea is the characteristic feature - Cognitive symptoms may occur at or before the onset of motor symptoms |
|
|
What are symptoms of Huntington Disease that may not be present immediately at onset?
|
- Many patients eventually become demented with disease progression
- Neuropsychiatric symptoms are present in 98% of patients (>10% have attempted suicide) |
|
|
How common is Huntington Disease? When is it more common?
|
- 4-8 / 100,000 people
- Symptoms present in 4th and 5th decade - Earlier age of onset in subsequent generations found when mutated gene passed to offspring from father |
|
|
How quickly does Huntington Disease progress?
|
Symptoms generally progress over several decades with complete motor disability and dementia approximately 20 years after symptom onset
|
|
|
When does anticipation occur in Huntington Disease? What does this mean?
|
When the mutated huntingtin gene on chromosome 4 is passed from the father to the offspring, there is an earlier age of onset in the subsequent generations, this is because of higher numbers of repeats of CAG
|
|
|
What is the pathogenesis of the huntingtin gene on chromosome 4 in Huntington Disease?
|
- CAG gene expansion causes a toxic gain of function in the huntingtin gene
- Mutation causes protein aggregation - Formation of intranuclear inclusions in the basal ganglia - Direct pathway to cellular injury is not understood |
|
|
What happens to the brain in Huntington Disease?
|
- Loss of medium striatal neurons in the caudate and putamen (these modulate motor activity)
- Loss results in increased motor ouput: chorea - Neuronal loss in cerebral cortex: cognitive changes |
|
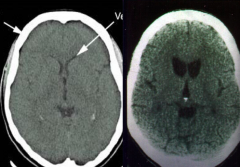
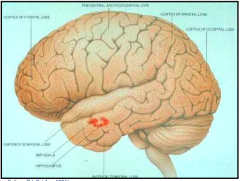
Which side is normal? Which is abnormal? What does the abnormal show?
|
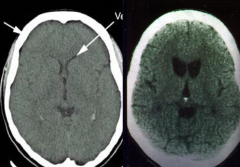
- Left: normal
- Right: Huntingtin Disease (atrophy of caudate and putamen w/ ventricular enlargement and mild to moderate atrophy of the gyri) |
|
|
What is the microscopic presentation of Huntington Disease?
|

Neuronal loss and astrocytosis in the caudate and putamen
|
|
|
What are the goals of therapy for Huntington Disease?
|
Treat the symptoms, not the disease
|
|
|
What types of drugs are used to treat Huntington Disease? Functions?
|
- Fluoxetine: depression
- Low dose anti-psychotics: delusions and paranoia - Tetrabenzine: inhibits VMAT for movement control |
|
Case 1:
- 78 yo presents with gradually worsening forgetfulness for 2 years - Impatience and irritability - Less interested in golf and other hobbies - Occasional difficulty finding words (defensive) - Family history: senility in father (in 70s) - Total Mini-Mental Status Exam: 26/30, short-term memory impairment, visual-spatial functioning impairment, pauses during word finding, impaired clock drawing test, and impaired verbal fluency - Fidgety, impatient, anxious, no evidence of psychosis or major depression - Labs: normal CBC, electrolytes, cholesterol; elevated TSH and low T4 What is the most likely diagnosis in this patient? |
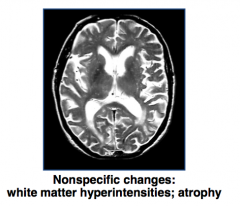
Alzheimer's Disease
|
|
|
Case 1:
- 78 yo presents with gradually worsening forgetfulness for 2 years - Impatience and irritability - Less interested in golf and other hobbies - Occasional difficulty finding words (defensive) - Family history: senility in father (in 70s) - Total Mini-Mental Status Exam: 26/30, short-term memory impairment, visual-spatial functioning impairment, pauses during word finding, impaired clock drawing test, and impaired verbal fluency - Fidgety, impatient, anxious, no evidence of psychosis or major depression - Labs: normal CBC, electrolytes, cholesterol; elevated TSH and low T4 What is considered a genetic risk factor for the development of this disease? |
Apolipoprotein ε4 allele
(risk factor for AD) |
|
|
Which of the following is an accurate explanation of the relationship between senile plaques and neurofibrillary tangles?
a) It is believed that a neurofibrillary tangle develops in a neuron followed by the neuritic plaque in the distal region of that neuron’s axon b) Their relationship is not understood at present c) They have no relationship as neurofibrillary tangles are incidental findings in the elderly and senile plaques are specific for AD d) They both occur in parallel and independently, in response to deposition of Aβ protein in the brain vessels (amyloid angiopathy) |
Their relationship is not understood at present
|
|
|
Which of the following abnormalities are required for a definitive diagnosis of Alzheimer’s disease?
a) A pathological analysis showing the presence of diffuse plaques and neuronal loss in multiple neocortical regions. b) A pathological analysis showing the presence of widespread neurofibrillary tangles in the brain. c) Clinical diagnosis of dementia & pathological analysis showing the presence of numerous neuritic plaques and neurofibrillary tangles d) Evidence of decreased brain activity in the frontal and parietal cortices on PET scanning e) The presence of elevated tau and Aβ proteins in the cerebrospinal fluid |
Clinical diagnosis of dementia & pathological analysis showing the presence of numerous neuritic plaques and neurofibrillary tangles
|
|
|
Case 1:
- 78 yo presents with gradually worsening forgetfulness for 2 years - Impatience and irritability - Less interested in golf and other hobbies - Occasional difficulty finding words (defensive) - Family history: senility in father (in 70s) - Total Mini-Mental Status Exam: 26/30, short-term memory impairment, visual-spatial functioning impairment, pauses during word finding, impaired clock drawing test, and impaired verbal fluency - Fidgety, impatient, anxious, no evidence of psychosis or major depression - Labs: normal CBC, electrolytes, cholesterol; elevated TSH and low T4 Which of the following is correct regarding treatment for this patient for his impairment? a) No treatment is indicated b) Prescribe a centrally-acting cholinesterase inhibitor; tacrine is the best c) Prescibe Memantine, an NMDA channel blocker d) Prescribe L-DOPA and carbidopa in anticipation of development of parkinsonsism e) Prescribe ropinerole, a selective D2 agonist |
Prescibe Memantine, an NMDA channel blocker
|
|
|
Case 2:
- 75 yo right handed man w/ 1-yr hx of progressive short term memory loss - Admitted with "acute delirium" - Waxing and waning confusion - Restless and wanders at night - Hallucinations (saw people in house) - Mini Mental Status Exam: 18/30 w/ deficits in orientation, calculations, word recall, and copying intersecting pentagons - Neuro exam shows bradykinesia, mild cogwheeling rigidity, no resting tremor, slow gain w/ decreased arm swing Given the described clinical presentation and course, which of the following is the best description of the expected findings at autopsy? a) Cytoplasmic accumulations of abnormal tau in the neurons of the frontal and temporal lobes b) Lewy bodies in the cerebral cortex and substantia nigra c) Neuronal loss and neurofibrillary tangles in the substantia nigra d) Numerous neuritc plaques and neurofibrillary tangles in the cerebral cortex e) TDP-43 deposits in the frontal and temporal lobes |
Lewy bodies in the cerebral cortex and substantia nigra
|
|
|
Case 2:
- 75 yo right handed man w/ 1-yr hx of progressive short term memory loss - Admitted with "acute delirium" - Waxing and waning confusion - Restless and wanders at night - Hallucinations (saw people in house) - Mini Mental Status Exam: 18/30 w/ deficits in orientation, calculations, word recall, and copying intersecting pentagons - Neuro exam shows bradykinesia, mild cogwheeling rigidity, no resting tremor, slow gain w/ decreased arm swing Which of the following statements is/are correct regarding the relationship of this patient’s disease with Parkinson’s disease? a) Both are always associated with the pathological findings of Alzheimer’s Disease b) They are completely unrelated to each other. c) They both exist on a spectrum of diseases all associated with alpha-synuclein deposition d) This patient’s disease differs from Parkinson’s disease because most cases are associated with a mutation in the alpha-synuclein gene and it is inherited in an autosomal dominant manner. |
They both exist on a spectrum of diseases all associated with alpha-synuclein deposition
|
|
|
Case 2:
- 75 yo right handed man w/ 1-yr hx of progressive short term memory loss - Admitted with "acute delirium" - Waxing and waning confusion - Restless and wanders at night - Hallucinations (saw people in house) - Mini Mental Status Exam: 18/30 w/ deficits in orientation, calculations, word recall, and copying intersecting pentagons - Neuro exam shows bradykinesia, mild cogwheeling rigidity, no resting tremor, slow gain w/ decreased arm swing Which of the following medication would be the best initial treatment for this patient? a) Riluzole b) Amantadine c) Selegiline d) Rivastigmine e) Baclofen |
Rivastigmine (centrally acting cholinesterase inhibitor, used for AD, Dementia with Lewy Bodies, and Vascular Dementia)
|
|
|
Case 3:
- 57 yo right handed man referred for marked progressive apathy - Sees a psychiatrist for >10 years for major depressive episode - Maintained ADLs at 45, but now declining in activity levels - Withdrawn from almost all activities except basketball on TV - Severely apathetic and hypersomnolent (sleep ~15 hrs/day) - Denies sadness, hopelessness, or worthlessness - Became much more passive than his original personality - Prior treatment with SSRI had no effect What is the best clinical diagnosis of his condition? |
Frontotemporal Dementia / Lobe Degeneration
|
|
|
Case 3:
- 57 yo right handed man referred for marked progressive apathy - Sees a psychiatrist for >10 years for major depressive episode - Maintained ADLs at 45, but now declining in activity levels - Withdrawn from almost all activities except basketball on TV - Severely apathetic and hypersomnolent (sleep ~15 hrs/day) - Denies sadness, hopelessness, or worthlessness - Became much more passive than his original personality - Prior treatment with SSRI had no effect Given the young age of onset of this patient's symptoms, what additional history would be important to elicit from his family members? a) Ages at which developmental milestones were achieved b) Exposure to heavy metals as a child c) Family history of similar symptoms in relatives d) History of febrile seizures e) Occupational exposure to hydrocarbons |
Family history of similar symptoms in relatives
|
|
|
Case 4:
- 54 yo man presents w/ 12 month history of R hand tremor, it is interfering with his daily activities - Tremor is worse when he is speaking or stressed, but if he concentrates it stops - Hand writing has changed - Gait has changed and he seems slower - Thrashes in bed and seems to be acting out his dreams - Mild decrease in facial expression w/ 6/sec rest tremor of R hand - Mild cogwheel rigidity in both upper extremities - Decreased arm swing in RUE - Muscle stretch reflexes decreased throughout What is the most likely diagnosis? |
Parkinson's Disease
|
|
|
Case 4:
- 54 yo man presents w/ 12 month history of R hand tremor, it is interfering with his daily activities - Tremor is worse when he is speaking or stressed, but if he concentrates it stops - Hand writing has changed - Gait has changed and he seems slower - Thrashes in bed and seems to be acting out his dreams - Mild decrease in facial expression w/ 6/sec rest tremor of R hand - Mild cogwheel rigidity in both upper extremities - Decreased arm swing in RUE - Muscle stretch reflexes decreased throughout Which of the following is the best initial treatment for this patient? a) Amantadine b) Apomorphine c) L-DOPA d) Pramipexole e) Selegiline f) Tolcapone |
Pramipexole (direct dopamine agonist, initial therapy in young patients)
|
|
|
Case 4:
- 54 yo man presents w/ 12 month history of R hand tremor, it is interfering with his daily activities - Tremor is worse when he is speaking or stressed, but if he concentrates it stops - Hand writing has changed - Gait has changed and he seems slower - Thrashes in bed and seems to be acting out his dreams - Mild decrease in facial expression w/ 6/sec rest tremor of R hand - Mild cogwheel rigidity in both upper extremities - Decreased arm swing in RUE - Muscle stretch reflexes decreased throughout You decide to treat the patient with pramipexole. Which of the following is an adverse effect of this drug? a) Insomnia b) Confusion c) Blood dyscrasias d) Seizures e) Hypertension |
Confusion
|
|
|
Case 5:
- 44 yo woman presents w/ difficulty walking and falls - She has had jittery feet for about 5 years, which progressed to involve all of her limbs and trunk - She is occasionally irritable and has poor concentration - Father died at 70 after several years of "funny movements" - Severely impaired eye movements, moderate chorea in all four limbs, wide based gait frequently interrupted by choreic movements of legs In this disease which of the following accurately describes the disease-causing mutation? a) Deposition of Aβ in the brain results from gain of function mutation b) Expanded trinucleotide repeats in CAGs c) Insertion mutation in a promoter gene d) Point mutation resulting in loss of protein function e) Point mutation resulting in decreased dopamine production |
Expanded trinucleotide repeats in CAGs
|
|
|
Case 5:
- 44 yo woman presents w/ difficulty walking and falls - She has had jittery feet for about 5 years, which progressed to involve all of her limbs and trunk - She is occasionally irritable and has poor concentration - Father died at 70 after several years of "funny movements" - Severely impaired eye movements, moderate chorea in all four limbs, wide based gait frequently interrupted by choreic movements of legs In Huntington’s disease, which of the following is the best explanation of the presence of chorea in this disease? a) Decreased cholinergic signaling to the cerebral cortex b) Increased dopamine release to the striatum c) Increased inhibition of the subthalamic nucleus d) Primary degeneration of the thalamus |
Increased inhibition of the subthalamic nucleus
|