Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
74 Cards in this Set
- Front
- Back
- 3rd side (hint)
|
Chr14: presenilin 1
Chr1: presenilin 2 Chr21: APP These 3 genes are all involved in early onset of this disease. |

AD
|
|
|
|
cortex: atrophy, degeneration of neurons
hippocampus: memory deficits basal nucleus of meynert & basal forebrain: degeneration of basal forebrain cholinergic neurons |

AD
|
|
|
|
progressive dementia:
1. progressive decline in short-term memory 2. lang/speech difficulties 3. visuospatial disorientation 4. apraxia 5. personality changes |

AD
|
|
|
|
-Most common cause of dementia in US.
-Imparied memory, cognition, functional decline. -Equal: men/women -50% of 85+ YO -Down's (trisomy 21) is risk factor |

AD
|
|
|
|
Large amts. of senile/neuritic plaques (with amyloid beta) and neurofibrillary tangles (with large amts. of phosphorylated tau protein) are necessary for diagnosis of what disease?
|

AD
|
|
|
|
cortical atrophy (same as AD)
|

DLB
|
|
|
|
1. dementia
2. parkinsonisms: bradykinesia, tremor, resistant to L-DOPA, hallucinations Dementia + Hallucinations=DLB 3. cognitive changes different than those seen in PD |

DLB
|
|
|
|
What abnormal protein deposits are found in the cortex, basal ganglion, & substantia nigra--in what disease?
|

LB's, DLB
|
|
|
|
What abnormal protein contains deposits of alpha-synuclein & ubiquitin?
|
LB
|
|
|
|
In what brain structure are LB's well defined? What disease process does this involve?
|

Substantia Nigra, DLB
|
|
|
|
In what brain structure are LB's ill defined? What disease process does this involve?
|

Cortex, DLB
|
|
|
|
What disease causes progressive dementia, with an unknown cause?
|
FTD
|
|
|
|
-No plaques, tangles, or lewy bodies
-lesions in frontal/temporal cortex-->"blade-like gyri" |
FTD
|
|
|
|
-Non-Alzheimer's Dementia, affects Frontal and Temporal Lobes
-Personality change, language dysfunction, and affective symptoms |
FTD
|
|
|
|
1. Loss of neurons
2. Extreme atrophy of frontal and temporal lobes 3. Tau Protiens inclusions |
FTD
|
|
|
|
Variant of FTD
|
Pick's Disease (Lobar Atrophy)
|
|
|
|
Extreme Atrophy limited to frontal and temporal lobes of brain.
-Neurons distend/stain + for ubiquitin inclusions |
Pick's Disease (Lobar Atrophy)
|
|
|
|
-Cerebral atrophy with gliosis and loss of cortical neurons, especially affecting temporal and frontal lobes.
-rare -onset~60 YO -ubiquitin marks the abnormal proteins for destruction |
Pick's Disease (Lobar Atrophy)
|
|
|
|
What are round/ovoid intraneural cytoplasmic inclusions containg p-lated tau called? What diesease process are they involved in?
|
Pick Bodies, Pick's Disease (Lobar Atrophy)
|
|
|
|
A patient presents with dementia and no other specific features, what disease does he most likely have?
|

AD
|
|
|
|
Patient presents with dementia and hallucinations, what disease does the patient most likely have?
|

DLB
|
|
|
|
Patient with dementia and an autopsy report shows severe atrophy of the brain in the frontal and temporal regions. How did the patient die?
|
FTD
|
|
|
|
-expanded polygluatamine repeat in exon
-anticipation -autosomal dominant |
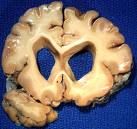
Huntington's Disease
|
|
|
|
-Atrophy of the Straitum (caudate and the putamen), cortex, globus pallidus
-Atrophy in caudate nucleus causes boxcar ventricles |
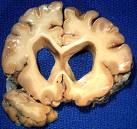
Huntington's Disease
|
|
|
|
Clinical presentation involves chorea, behavioral and cognitive changes
|
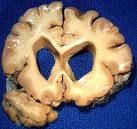
Huntington's Disease
|
|
|
|
1.Striatum Atrophy
2.Trinucleotide Repeat Disease 3.Neuronal Loss 4.Gliosis 5.NO Inclusion Bodies 6.Onset 35-40 YO |
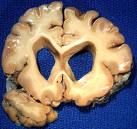
Huntington's Disease
|
|
|
|
-Synuclein protein, this is the major protein found in what? What disease is synuclein protein involved in?
|

Lewy Bodies, Parkinson's Disease
|
|
|
|
LB's which cause degeneration of the neurons in the substantia nigra-->decrease dopamine-->depigmentation
|

Parkinson's Disease
|
|
|
|
1.Resting Tremor
2.Bradykinesia 3.Rigidity 4.Gait Instability |

Parkinson's Disease
|
|
|
|
Microscopically-->see LB's which have a dense core and a clear halo.
|

Parkinson's Disease
|
|
|
|
-autosomal recessive
-decrease plasma binding of Cu -can be reversed by a Cu chelator |
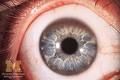
Wilson's Disease
|
|
|
|
Effects the basal ganglion, this is a disease of theliver and of the lens
|
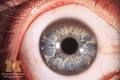
Wilson's Disease
|
|
|
|
-cornea has manifestation of Kayser-Fleischer Rings
-occurs in adolescents and young adults -movement disorders such as dystonia -behavioral, cognitive changes |
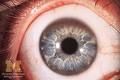
Wilson's Disease
|
|
|
|
Copper deposits (toxic levels) in: liver, cornea, basal ganglia (lenticular nuclear=putamen + globus pallidus)
|
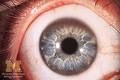
Wilson's Disease
|
|
|
|
Chr21 that affects copper/zinc superoxide dismutase gene
|

ALS
|
|
|
|
Skeletal muscle atrophy, lateral column degeneration due to corticospinal tract degeneration, atrophies of the spinal roots
|

ALS
|
|
|
|
-Rapidly progessive UMN & LMN failure leading to death, most often respiratory failure
-UMN-->increased reflexes -LMN-->fasiculations |

ALS
|
|
|
|
-UMN degeneration-->cortical atrophy-->degeneration of the lateral cortical spinal tract
-LMN degeneration-->anterior motor neurons of spinal cord -skeletal muscle atrophy |

ALS
|
|
|
|
-Autosomal recessive trinucleotide repeat in intron-->affects regulation of gene expression
|

Friedreich's Ataxia
|
|
|
|
Lesions occur in:
1.DRG 2.Spinal Cord 3.Cerebellum |

Friedreich's Ataxia
|
|
|
|
1.Gait Ataxia
2.Cerebellar Incoordination 3.Sensory Deficits 4.Cardiac & other abnormalities |

Friedreich's Ataxia
|
|
|
|
Onset ~10 YO (before 25 YO typically)
|

Friedrich's Ataxia
|
|
|
|
-protein with abnormal configuration
-replicates by transforming a normal protein into an abnormal conformation -are resitant to degradation -accumulate in specific cell types-->cause disease b/c of neuronal cell death |
Prion Disease
|
|
|
|
-Characteristics of Disease: Progressive neurological deterioration, heterogenous phenotype for a variety of reasons.
|
Prion Disease
|
|
|
|
-Normal conformation of protein is an alpha-helix
-Abnormal conformation of protein=B-pleated sheets which are protease resistant and form aggregates |
Prion Disease
|
|
|
|
Codon at 129: Met/Val
Codon at 178: Asp |
Normal Prion Protein
|
|
|
|
Codon at 129: Val
Codon at 178: Asn |
CJD with cortex lesions
|
|
|
|
Codon at 129: Meth
Codon at 178: Asn |
-CJD with lesions to thalamus & superior olive
-Pt presents with Fatal Familial Insomnia |
|
|
|
-Clinical course is dementia with myoclonus and progressive neurological decline.
-Death usually occurs in less than a year after onset of symptoms. |
CJD
|
|
|
|
-Pathological changes include spongiform encephalopathy.
-Also neural necrosis |
CJD
|
|
|
|
-Usually under 45 yo
-Behavioral changes more prominent and ataxia presents early on -All had characteristics of codon 129 which made them more susceptible |

Variant CJD in Great Britain, AKA Mad Cow Disease
|
|
|
|
An infection of Bovine Spongiform Encephalopathy causes what disease?
|

Variant CJD in Great Britain, AKA Mad Cow Disease
|
|
|
|
-CNS lesion: cerebral & cerebellar cortex
-Motor nerves affected -Children: CNS & PNS -Adults: PNS |

Lead Poisoning
|
|
|
|
CNS: edema, white matter necrosis, vascular proliferation, glial proliferation, neuron damage
PNS: segmental demyelination & axon degeneration |

Lead poisoning
|
|
|
|
CNS effects (esp children):
-acute-->increased ICP, seizure -chronic-->cognitive effects PNS: segmental demyelination of motor nerves-->wrist/foot drop, slowed nerve conduction, later axonal degeneration |

Lead poisoning
|
|
|
|
Results primarily in sensory neuropathy
|
Arsenic Poisoning
|
arSENic poisoning results in SENsory neuropathy
|
|
|
Permanent deficits in 4 locations:
1.Cerebral cortex (laminar necrosis) 2.Hippocampus (pyramidal cells) 3.Cerebellum (purkinje cell death) 4.Globus Pallidus necrosis *** (Globus Pallidus shows discoloration signaling a movement disorder) |

CO toxicity
|
|
|
|
Etiology is hypoxia
|
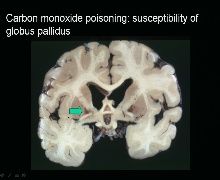
CO toxicity
|
|
|
|
Symtoms: HA, dizzy, confusion
|
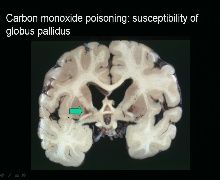
CO toxicity
|
|
|
|
Lesion Location:
-Anterior horn -Posterior columns |

B1/thiamine deficiency: alcoholic polyneuropathy
|
|
|
|
Etiology:
-Due to thiamine deficiency-->bad nutrition |

B1/thiamine deficiency: alcoholic polyneuropathy
|
|
|
|
-Myelin and axon degeneration in anterior horn
-posterior root degeneration & 2ndary degeneration of posterior columns |

B1/thiamine deficiency: alcoholic polyneuropathy
|
|
|
|
Symptoms:
-numbness, paresthesias, weakness -motor and sensory weakness, mostly distal |

B1/thiamine deficiency: alcoholic polyneuropathy
|
|
|
|
confusion, opthalmoplegia, and ataxia
This classic triad of symptoms occurs in what illness |

Wernicke's Encephalopathy
|
|
|
|
What occurs first, WE or KP?
|

WE
|
|
|
|
1.Is WE or KP reversible if given thiamine?
|

WE
|
|
|
|
-irreversible, causing memory disturbance and confabulation
|

KP
|
|
|
|
Lesion location:
1. periaqueductal gray matter including third nerve nuclei and MLF-->causes opthalmoplegia 2. floor of 4th ventricle including the vestibular nucleus-->causes ataxia |

WP
|
|
|
|
Lesion location:
-mamillary bodies, medial dorsal thalamus-->cause memory deficits and cognitive symptoms -mamillary body lesions look red b/c of hemorrhage |

KP
|
|
|
|
-small petechial hemorrhages in mamillary bodies
-vascular changes: endothelial prominence & petechial hemorrhages -neuronal necrosis -macrophage response & gliosis |

B1/thiamine deficiency: WE & KP
|
|
|
|
Lesion Location:
-spinal cord-thoracic (primary myelin sheaths-->degen axons)-->reversible -dorsal columns-->involved early -lateral columns-->corticospinal tract, spinothalamic tract-->contributes leg involvement |
Vit B12 deficiency
|
|
|
|
-Decreased intake, impaired absorption (IF def/pernicious anemia)
-Folic acid: releive anemia (not neuro def) |
Vit B12 def
|
|
|
|
-Motor & sensory degeneration
-Ataxia-->caused by sensory deficits or cerebellar deficits -Sensory def (numb/tingling) -Loss of vibration, proprioception, pain -LE weakens (due to thoracic spinal cord) -Increased deep tendon reflexes/Babinski signs -Fatigue |
Vit B12 deficiency
|
|
|
|
-Associated with elevated blood ammonia levels-->cause toxicity in brain
-Symptoms: Disturbances in consciousness, motor abnormalities |
Hepatic Encephalopathy
|
|