Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
84 Cards in this Set
- Front
- Back
|
what’s the effect of Ach esterase inhibitors?
|
inhibitors of acetylcholine esterase will potentiate the effects of ACh
|
|
|
what are some other, more dangerous inhibitors of ACh esterase? what’s their common chemical property?
|
other inhibitors of ACh esterase include nerve gases like tabun and sarin, and an insecticide parathion. the common chemical property is that they are all phosphate esters with an “active ester bond,” P-CN, P-F, etc. that are highly polar and very susceptible to hydrolysis
|
|
|
what’s the mechanism for parathion, tabun and sarin? what are the effects on ACh?
|
when the insecticide or nerve gases gain access to the neuromuscular junction, the ACh esterase will hydrolyze it and you get a phospho-serine intermediate, which is irreversible, so it’s like a suicide reagent for the enzyme. it potentiates the effects of ACh in an uncontrolled way
|
|
|
what is PAM? how does it work?
|
pyridine aldoximine methiodide (PAM), an older drug, an antidote for nerve gas poisoning. the pyridinium group orients in the anonic site and the monoxine moiety hydrolyzes off the phosphate ester to regenerate active enzyme
|
|
|
soldiers are given muscarinic antagonists. what do they protect against?
|
muscarinic antagonists to protect the SNS from overstimulation
|
|
|
what are the 4 steps of ACh synthesis up to release?
|
1) single condensation reaction generates ACh in the cytoplasm, 2) the enzyme choline acetyltransferase catalyzes the rxn (CAT - located uniquely in cholinergic neurons), 3) the ACh is taken up into the vesicles, concentrated there, 4) released upon depolarization
|
|
|
what are the 3 steps following ACh release?
|
1) where at the post-synaptic site, it interacts with the cognate receptors: nicotinic and muscarinic, which have different functions and localizations in the CNS and periphery. 2) ACh is rapidly hydrolyzed by choline esterase, generating acetate and choline, 3) the latter of which is taken up into the nerve terminal through a specific transporter to be used in de novo synthesis.
|
|
|
what’s the key step for regulation of ACh levels?
|
Reuptake of choline is key to regulation of ACh levels –Choline is rate limiting in ACh biosynthetic pathway; it’s a well-regulated system even at the level of the individual neuron
|
|
|
where are cholinergic neurons found?
|
you should know all motor neurons are cholinergic, cell bodies in the ventral horn, projections etc.
|
|
|
what degenerates in Huntington disease?
|
striatal interneurons degenerate in Huntington disease
|
|
|
what’s one thing that degenerates in the brains of people with Alzheimer’s disease? and what has been an attempted therapeutic strategy?
|
cell bodies in the nucleus basalis* with rather diffuse projections are important because they seem to degenerate early in the brains of people with Alzheimer’s disease, so some of the therapeutic strategies had been to increase cholinergic transmission in early-onset patients to help with cognitive function
|
|
|
what do congenital myasthenic syndromes affect? what happens with neurotransmission?
|
Presynaptic Function. would all reduce neurotransmission at the neuromuscular junction, translating into muscle weakness but very rare (e.g. Choline acetyltransferase (CAT) deficiencies reduce the amount of ACh available for release (6 cases); Paucity of presynaptic vesicles (1 case), may be embryonically lethal
|
|
|
what’s Myasthenia gravis? what’s a hallmark feature?
|
Myasthenia gravis is a neuromuscular disorder characterized by weakness of skeletal muscle. hallmark feature: Ptosis with eyebrows up
|
|
|
what are 7 common features of myasthenia gravis?
|
Common features of MG: 1) Only the motor system is impaired, 2) Likely to affect cranial nerves and limb muscles, 3) Fatigability of muscle upon use*, 4) Severity of disease can vary dramatically during course of a single day. 5) No clinical signs of denervation which would lead to muscle atrophy and loss of reflexes but 6) reflexes intact and 7) weakness can be reversed by acetylcholinesterase inhibitors
|
|
|
what do you see on electromyleogram (EMG) with MG patients?
|
Characteristic abnormal electromyleogram (EMG) seen with MG patients, the fast-stem muscle stimulation decline/decrement
|
|
|
what is the hypothesis explaining MG? what type of disease is it?
|
rabbit gave rise to the MG hypothesis that the production of auto-antibodies against the ACh nicotinic receptor as cause of the disease, (best evidence is when injecting antibodies from people with MG and inject into animal model causing disease, “passive transfer”)
|
|
|
what 2 structural changes do you see at the neuromuscular junctions of patients with myasthenia gravis?
|
Reduced AChR sites at the neuromuscular junctions of myasthenia patients, by 90% in biopsies; 2) can also see damaged synapses (probably due to inflammation), certainly part of the story
|
|
|
what happens to antibodies in patients with MG? what are the consequences?
|
1) if normal turnover is 5-7 days, in MG, antibodies cross-link; they are bivalent and this will increase turnover as receptors are aggregated by antibodies and then go through lysosomal destruction --> decline in receptor number (measured clinically in patients);
|
|
|
what’s the overall goal of treatment in MG? what agents are used?
|
to enhance transmission across the synapse. cholinesterase inhibitors are effective because whatever receptors are present, the ACh will be increased and its action will be prolonged at the synapse, which seems to help. Need to worry about immunological involvement as well, so immunosuppressants are often prescribed, and ways to reduce inflammation (steroid therapy) (remember case, thymectomy, plasmaphoresesis 2x/wk)
|
|
|
what’s Lambert-Eaton syndrome? compare and contrast with MG.
|
Lambert-Eaton syndrome: Rare myasthenic disease also involving proximal muscle weakness – most characteristic but weakness seen in RESTING muscle and improves with use. Different from MG patients who tend to have leg weakness and difficulty walking.
|
|
|
what’s the muscle response in Lambert-Eaton syndrome? what ion is involved?
|
Very different muscle response than seen with MG patients (like opposite), called facilitation with fast rep. stimulation. calcium tends to accumulate and you will then get a more normal type of release. Repeated stimulation leads to Ca accumulation in presynaptic terminal, restoring normal release
|
|
|
what are some symptoms you see in Lambert-Eaton syndrome? what’s the etiology in many patients?
|
In Lambert-Eaton syndrome, you see autonomic dysfunction e.g. dry mouth, dry eyes, constipation. In a significant fraction of patients, it’s a paraneoplastic disorder, meaning it can be associated with small cell lung cancer tumors. It, too, is an autoimmune disease.
|
|
|
how do scientists show that a disease is autoimmune?
|
for autoimmune diseases, you can passively transfer disease to mice via antibodies from people with the disease. unknown why certain people start making these antibodies and others don’t
|
|
|
people with Lambert-Eaton syndrome have high titers of antibodies against the ____ channel
|
people with Lambert-Eaton syndrome have high titers of antibodies against the calcium channel
|
|
|
what happens with calcium channels in Lambert-Eaton syndrome?
|
by blocking the amount of calcium entering the cell with the autoantibodies, it causes dysfunction. as the neuron depolarizes, normally you get calcium influx and release, but this doesn’t happen properly.
|
|
|
Why would tumors lead to anti-Ca channel antibody production?
|
in the small-cell lung carcinomas, some have calcium channels on their surface, so immune system is being presented these calcium channels in an abnormal way, so the immune system in these patients make antibodies in response
|
|
|
what’s the overall strategy for treating Lambert-Eaton syndrome?
|
you need to enhance calcium current, can do so with potassium channel blockers, because K+ efflux is a repolarizing event for the membrane so if you block repolarization, you’ll have longer depolarization and thus increased calcium influx (e.g. 3,4-DAP); non-used muscle strength being restored. choline esterase inhibitors don’t work… “that’s what we see”
|
|
|
contrast the 2 types of Myasthenic acquired diseases.
|
2 types of Myasthenic acquired diseases: post-synaptic (MG on ACh nicotinic receptors) vs. pre-synaptic effect (Lambert-Eaton syndrome on calcium channels)
|
|
|
what are the 2 famous catecholamines? what’s the main structural difference?
|
dopamine -> dopaminergic; norepinephrine/ noradrenaline -> noradrenergic (hydroxyl group at the beta position)
|
|
|
what’s the precursor for all catecholamines?
|
Tyrosine serves as the precursor taken up from the circulation for all of the catecholamines.
|
|
|
what’s the first step in the dopamine synthesis pathway?
|
the first step is hydroxylation of tyrosine in meta position, catalyzed by Tyrosine hydroxylase, and the product is L-dopa
|
|
|
what’s a marker protein for dopaminergic neurons?
|
The enzyme (tyrosine hydroxylase) is uniquely localized in catecholaminergic neurons, located in the cytoplasm and highly localized in the nerve terminals of neurons (just like cholinergic), the major site of biosynthesis is at the nerve terminals itself
|
|
|
what’s involved in the rate-limiting step in dopamine synthesis?
|
Tyrosine hydroxylase is involved in the rate-limiting step, the regulatory step here; it’s activated by calcium and cAMP and other second messengers
|
|
|
what controls the local rate of dopamine synthesis?
|
end-product inhibition; negative feedback. there’s actually allosteric binding site on the enzyme which is capable of recognizing the product, dopamine or norepinephrine. if there’s too much product being made, then you get this feedback effect. this is important for the local rate of synthesis
|
|
|
what inactivates tyrosine hydroxylase? reversible or irreversible? why important?
|
Norepinephrine binds tyrosine and forms an inactive complex, which is reversible. this regulatory feedback is key to keeping everything in balance, to keeping a steady pool of neurotransmitters, enough there to be released but not too much!
|
|
|
in what scenario would tyrosine hydroxylase be inactivated?
|
during periods of relatively low activity, NE will tend to accumulate in the nerve terminal, tyrosine hydroxylase will be converted into an inactive form
|
|
|
when would tyrosine hydroxylase levels become active? slow or fast?
|
if you go to a high-impulse flow condition and NE levels decline as it’s being released in the nerve terminal. tyrosine hydroxylase, by mass action, will tend to be liberated and you go from an inactive to an active form; this can happen very quickly!
|
|
|
if you had to make more tyrosine hydroxylase rather than activate it, would the process be slow or fast?
|
if you had to make more enzyme (which occurs up in the cell body and is transported down to the nerve terminal, etc.), there’s no way to have a regulatory process that will quickly respond to changes in impulse flow like this.
|
|
|
what are autoreceptors?
|
autoreceptors are presynaptic receptors activated by the transmitter released from the same synapse (found in D2 receptors).
|
|
|
what’s the role of autoreceptors in the dopaminergic biosynthetic pathway?
|
they regulate tyrosine hydroxylase. autoreceptors are involved in negative-feedback loop mediated through activation of presynaptic receptors.
|
|
|
what happens if dopamine levels rise to too high a level?
|
normally, dopamine will act at its post-synaptic site, but if dopamine levels rise to a level that’s too high, there’s negative feedback. you begin to activate autoreceptors which (through signaling) dampen tyrosine hydroxylase activity at the local synapse.*
|
|
|
what happens to L-dopa?
|
L-dopa is not around very long. it’s rapidly converted into dopamine by an enzyme called DOPA decarboxylase (a very active though not very specific enzyme)
|
|
|
what is L-dopa used for therapeutically? why?
|
in Parkinson’s patients where losing dopaminergic activity, L-dopa is used as a drug in early stages to compensate partially for that loss. by providing more of this direct precursor of dopamine you can improve neurological function.
|
|
|
where is dopamine made? where is it stored?
|
after dopamine is made in the cytosol, it is taken up into vesicles at the nerve terminals (like ACh)
|
|
|
what’s the final step of NE synthesis in noradrenergic nerve terminals? what enzyme is involved?
|
a final step in the noradrenergic nerve terminals is the hydroxylation at the beta position of DA by the enzyme DBH, dopamine-beta-hydroxylase.
|
|
|
where is DBH?
|
DBH is located in the synaptic vesicles themselves. the last step in the noradrenergic neurons does take place in those vesicles. dopamine is taken up and converted into norepinephrine there.
|
|
|
what’s a marker protein for noradrenergic neurons in the brain?
|
DBH can be used as a marker for noradrenergic innervation because it’s specific
|
|
|
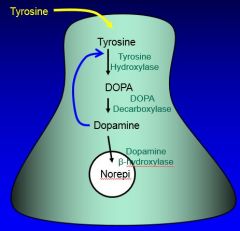
(norepi only in noradrenergic neurons)
|
|
|
what two enzymes are involved in the degradation of catecholamines?
|
there are 2 *important* catabolic processes for degradation of the catecholamines: Monamine oxidase (MAO) and Catecholamine-O-methyl transferase (COMT). Neither one of these enzymes is responsible for inactivation of synaptic activity** they are more catabolic, part of the steady-state turnover.
|
|
|
what’s MAO? what does it do?
|
Monamine oxidase (MAO), mitochondrial enzyme, takes any amine and converts into its corresponding aldehyde. kind of functions as a scavenger, will chew up a variety of amines, certainly catecholamines in the nerve terminal will be degraded into deaminated metabolites
|
|
|
what’s COMT? what does it do?
|
Catecholamine-O-methyl (COMT), the other enzyme, which adds a methyl group to catecholamines, taking it down a metabolic pathway toward degradation and ultimate excretion
|
|
|
what’s the effect on catecholamines if you inhibit MAO or COMT?
|
you can inhibit these enzymes, e.g. COMT, and you will NOT potentiate the effects of catecholamines at the nerve terminals (very different from ACh and acetylcholinesterase inhibitors)
|
|
|
what inactivates dopamine and what potentiates DA?
|
dopamine (DA) is inactivated by a specific transporter expressed on the pre-synaptic terminal that takes DA back up into the nerve terminal itself. this process is critical because inhibitors for the transporter will indeed potentiate the effects of DA;
|
|
|
what can potentiate the effects of NE?
|
similarly, inhibitors of the norepinephrine (NE) transporter will potentiate the effects of NE
|
|
|
what are 2 roles of transporter proteins?
|
1) the transporters are responsible for high-affinity reuptake and 2) they have the main role in inactivation / clearance.
|
|
|
what’s the gene family for transporters? what’s their structure? what’s a common feature?
|
transporter proteins are part of the SLC6 gene family. are large and complex. their structure: 12-transmembrane segments, assemble into dimers. all share common features e.g. use Na as a co-transporter to bring neurotransmitter back into the nerve terminal
|
|
|
what does the DAT transporter transport? what inhibits it(3)? useful in what diseases(2)?
|
the DAT transporter has endogenous substrates DA, NE, Epi. its inhibitors are benztropine, cocaine, amphetamines. drugs inhibiting the DAT transporter have been used in ADHD and Parkinson’s
|
|
|
what does the NET transporter transport? what inhibits it(2)? useful in what disease?
|
the NET transporter also has endogenous substrates DA, NE, Epi. its inhibitors are nortriptyline and desipramine, which have been used to treat depression
|
|
|
what does the 5-HT transporter transport? what inhibits it (3)? useful in what diseases (3)?
|
the 5-HT transporter transports serotonin. its inhibitors include fluoxetine, MDMA (ecstasy), and escitalopram. therapeutic uses for inhibitors include treatment of OCD, anxiety, and depression
|
|
|
there’s a long-term link between catecholamines and what disease?
|
there’s a long-term link between catecholamines and depression
|
|
|
if inhibiting SLC6 gene family transporters is useful for treating disease, what does this tell us about the molecular basis for pathophysiology?
|
that some disorders may be due to mutations of various SLC6 gene family transporters or other parts of the pathway. understanding their pharmacology has considerable clinical utility
|
|
|
what's the overall strategy in the metabolism of dopamine?
|
catechols can be methylated and can be oxidized, giving rise to various adducts. can occur in either order and in either way.
|
|
|
what can happen to aldehydes produced by MAO?
|
the aldehydes produced by MAO can either be reduced to an alcohol or oxidized to an acid
|
|
|
what generates HVA? why important?
|
in the case of dopamine, oxidation to DOPAC and ultimately methylation generates homovanillic acid (HVA). so measuring HVA levels in CSF is a diagnostic signal for dopaminergic activity**
|
|
|
when are HVA levels used clinically? what’s an example?
|
particularly important in “disorders of basal ganglia. for example, for Parkinsonian disorders, where your patient is losing dopaminergic activity, HVA levels are depressed in the CSF. with treatment, HVA levels will change. (no good signal in the periphery)
|
|
|
what’s a useful clinical assay for NE? what’s a disease it’s used for?
|
in the case of norepinephrine, the most useful assay has come from the periphery where oxidation of the aldehyde occurs to the corresponding acid, VMA (vanillylmandelic acid), which is a very useful diagnostic signal for norepinephrine in the periphery. e.g. in pheochromocytoma (NE-producing tumors in the adrenal gland), VMA levels in the urine are highly elevated. (there’s no good signal in the brain, so no useful diagnostic assay in that case)
|
|
|
what’s the structure of the adrenergic receptors?
|
for all of the adrenergic system, the adrenergic receptors have the classic 7 transmembrane sequence*, a motif that binds and activates G proteins.
|
|
|
what are the 2 main families of noradrenergic receptors?
|
Noradrenergic receptors, 2 main families: α receptors and β receptors. various subtypes
|
|
|
dopamine receptors are important in what disease?
|
dopamine receptors are important in schizophrenia.
|
|
|
compare and contrast DA-1 receptors vs. DA-2 receptors
|
localized in different areas of the striatum. DA-2 receptor found on corticostriatum afferents vs. DA-1 receptor found on interneurons in the striatum. also, DA-1 is linked to adenylate cyclase and regulated by guanine nucleotides.
|
|
|
what’s the effector of D1 receptors? what’s the effect of bromocriptine on it?
|
D1 receptors effector is cAMP, bromocriptine (a small-molecule alkaloid) is an antagonist, chromosome 5
|
|
|
where are D2 receptors found? what’s the effect of bromocriptine on them? the consequences?
|
D2 receptors are found in the pituitary, and if activated, they inhibit the release of prolactin** bromocriptine is an agonist. (chromosome 11)
|
|
|
which dopamine receptors have autoreceptors?
|
D2
|
|
|
neuroleptic (anti-psychotic) drugs are potent antagonists of what type of receptors?
|
neuroleptic (anti-psychotic) drugs are potent antagonists D2 receptors.
|
|
|
where in the brain is DA? (5)
|
dopamine has rather distinct localizations to the substantia nigra, striatum, olfactory “tubercle,” the frontal cortex and median eminence
|
|
|
which catecholamine is more diffusely localized in the brain?
|
NE is more diffusely localized throughout the brain. different from dopamine.
|
|
|
what are marker proteins (e.g. DBH) used for?
|
amino fluorescence and marker proteins used to map pathways, creating an “antibody sandwich” e.g. using tyrosine hydroxylase as a marker for dopaminergic pathways, DBH as a marker for noradrenergic receptors
|
|
|
what’s a very important pathway for dopamine that you should know?
|
should know the classic nigrostriatal pathway* for dopamine. it has cell bodies in the substantia nigra and ending up in the striatum (And Reha said “caudate putamen”),
|
|
|
what pathway degenerates in Parkinson’s disease?
|
the nigrostriatal pathway degenerates in Parkinson’s disease** this pathway connects different parts of the basal ganglia, which are involved in the coordination of motor movement. the degeneration of the pathway accounts for the “interesting spasticity and motor movement you see in patients with Parkinson’s.
|
|
|
what’s another important dopaminergic pathway? where are its cell bodies?
|
the other one is the mesolimbic pathway, also with cell bodies in the substantia nigra with projections up through the medial forebrain bundle into the olfactory tubercle, nucleus accumbens and the frontal cortex. this accounts for the localized distribution of dopamine there.
|
|
|
NE-producing neurons projection from what region? terminate where?
|
rather diffuse projections from the locus coeruleus throughout the brain terminate in spinal cord, brainstem, basal ganglia and entire isocortex. less active during periods of tonic vegetative behavior
|
|
|
why is Parkinson’s disease so important?
|
Parkinson’s Disease, is the classic example of the relationship between catecholamines and disease. you see a loss of critical dopaminergic pathway in the basal ganglia, which is why why L-dopa is a good drug because at least in early stage Parkinson’s, helps compensate for loss of dopamine with neurodegeneration. also why HVA is depressed because losing a major dopaminergic system in the brain.
|
|
|
what’s the catecholamine hypothesis of affective disorder?
|
basically posits that low levels of dopamine may explain depression and elevated levels may explain mania in bipolar disorder but turns out the disease are more complicated. e.g. serotonin is also important.
|
|
|
what’s the therapeutic strategy based on the catecholamine hypothesis?
|
to give drugs which elevate catecholamine levels, e.g. MOA inhibitors and transport inhibitors, for treatment of depression, which seem to be effective (at least for some)
|