Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
55 Cards in this Set
- Front
- Back
|
Protein Malnutrition:
Kwashiorkor syndrome Famine Edema Protein energy Malnutriton In US can occur in |
- protein deficiency but adequate calories
-edema gives puffy appearance -Inadequate syntehsis of plasma proteins especially albumin. -Osmotic pressure not maintained and fluid escapes into tissues -Water in EC space is increased relative to body weight -lack protein not calories -Starvation, lacks calories and proteins as well as other nutrients -pregnant and lactating women -eating disorders -elderly, chronic ill -chronic alcholoics/substance abusers -hospital patients -genetic disorders |
|
|
Essential vs. Non essental
|
-Essential = cannot synthesize
-Non essential = can synthesize via TCA PVT TIM HALL = Essential |
|
|
how much Protein do we need:
compared to fat/glucose? can we get all from one source? protein needs for: infants, children, teens, adults, pregnant, athletes kg/kg and g/day |
no significant storage pool, need to consume daily
proteins differ in contest of essential AAs and digestilbity, need multiple sources g/kg: Infants(2.2)>children(1.8-1.25)> atheletes(1.7-1.2)>teen(1.0-.8)> adults male and female(.8)[pregnant/lactating needs 20-30% more] G/day: Adult Male(56)> Teens(45-55)>female(44)> children(38-20)> infants(20-6.5) |
|
|
Nitrogen Balance Flow diagram
|
Dietary Protein-->digest-->aa:
-(translation)endogenous proteins -other N compounds -a-ketoacids: glucose, lipids, energy -NH3: urea-->excretion |
|
|
Nitrogen Balance
In N balance: Positive N balance: Negative N balance: |
excretion = intake
excretion<intake(growth, pregnaancy, tissue repair) excretion>intake( malnutrition, starvation, illness, surgery, burns) |
|
|
Pro-enzymes, converted by, active enzyme
pepsinogen trypsinogen chymotrypsinogen pro-carboxypeptidases pro-elastase |
pepsinogen-->H+ pepsinogen-->pepsin
trypsinogen-->enteropeptidase-->trypsin chymotrypsinogen-->trypsin-->chymotrypsin pro-carboxypeptidases-->trypsin-->carboxypeptidase pro-elastase-->trypsin-->elastase |
|
|
Problem with cystic fibrosis regarding enzymes?
|
-secretion is imparied, need supplement with pill
|
|
|
amino Acid absorption in small intestine is dictated by:
#, located, special, requries? |
6 brush-border enzymes specific for uptake of amino acids, for class of amino acids, requires energy
|
|
|
endogenous proteins back to amino acids is called?
used to? purpose? major sites of turn over? turn over time? |
Protein turnover
-dispose of damaged proteins and allow cells to respond to different conditions -conserves resource and reduces need to dispote of waste metabolic products -proteasomes(cytoplasm) and lysosomes -1/2 lifes of proteins vary |
|
|
Proteolysis in proteasomes process flow chart
where does UBQ bind? |
Protein + Ubquintin-->ubiquinylated protein
*ATP ubiquinylated protein-->proteosome-->peptides *ATP ubiquitin returned and not degraded -UBQ's carboxyl terminus forms an isopeptide bond with the E-amino groups of lysine in proteins. |
|
|
Proteasomes:
speed and specifity? for? structure(size, units, shapes)? how are proteins targeted? UBQ and degradation require? |
-fast and specific
-proteins of which must be regulated carefully and for damaged/mutant proteins -large, mutliunited, cylinder shaped witha central capped at both ends conjugation via Ubiqutin ATP |
|
|
Proteolysis in Lysosomes:
speed and specifity? for? suited for? |
-slow and non selective
-plasma membrane proteins and extracellular proteins taken in by endocytosis of phagocytosis -low pH |
|
|
Other proteolytic systems
|
-numberous other proteases in the cell requried for apoptosis(caspases)
|
|
|
Biosynthesis of non-essential amino acids
|
-glucose, lipids, energy-->a-ketoacids-->amino acids
|
|
|
Transamination
allows for? catalyzed by? |
transfer of a NH2 group from an amino acid to a ketoacid to make another amino acid
transaminases a-amino acid 1 + a-ketoacid 2 --> a ketoacid 1 + a-amino acid 2 |
|
|
a co-factor for reactions of amino acids other than transamination, and transaminases
derived from? conversion proces |
pyridoxal phosphate
Vitamin B6 pyridoxine(vitamin B6)-->pyridoxamine-->pyridoxal-->pyridoxal phosphate |
|
|
Key players in amino acid metabolism
|
glutamate
a-ketoglutarate glutamine |
|
|
Fxn of glutamate dehydrogenase rxn?
* |
converts glutamate into alpha ketolgutarate or a-ketoglutarate back to glutamate
* enables either the conversion of a-ketoglutarate-->glutatamine by donating at NH3(glutamate-->a-ketoglutarate as a consequence) *NAD is reduced or the reverse glutamine-->a-ketoglutarate by accepting NH3(also converting a-ketoglutarate(glutamate)--> glutamate as a consequence)) *NADPH is oxidized) |
|
|
synthesis of glutamine
need energy? |
-Glutamate-->glutamine(glutamine synthetase)
*ATP-->ADP, NH3 expended yes |
|
|
Hydrolysis of Glutamine
need energy? |
Glutamine--> glutamate
* H20-->NH2, glutaminase) no |
|
|
Fxn of glutamine
|
-non toxic form of transport and storage of ammonia-->most abundant amino acid in circulation
|
|
|
Synthesis of non-essential amino acids and role of pyridoxal phosphate
|
* = pyridoxal phosphate
glycolysis: glucose-->P-glycerate-->pyruvate: Phosphoglycerate-*>Serine-->gly/cys<--met Pyruvate-->ala From TCA: fumarate-->malate-->oxaloacetate: *->Asp-->Asn + Acetyl coA-->Citrate-->a-ketoglutarate: -->succinate-->fumarate-->malate -*-> Glu-->Gln+ Pro |
|
|
Synthesis of tyrosine:
derived from ____ via ____ cofactor: biopterin is syn from __ and is thus not a ___ rxn |
phenylalanine oxidation
tetrahydrobiopterin GTP, not a vitamine Phenylalanine-->Tyrosine * O2-->H20 *tetrahydrobipterin + NADH-->dihydrobiopterin-->NAD -DHB reductase - tetrahydrobipterin essentially donates an Hydrogen by forming a double bond |
|
|
Synthesis of Cysteine
2 steps? rxns require? which rxns act where? is homocystein essential? what is essentially happening |
1. Condesation of serine with homocysteine-->cystathionine
* cyastathionine snyhtase PLP lose H20 2. cleavage of cystathionin-->cysteine + a-ketobutyrate cystathionase, PLP, H20 condesation/cleavage occurs at different sides of the sulfur atom -no, not found in proteins -Sulfur group transfered from homocysteine to serine to form cystein |
|
|
Synthesis of cysteine
|
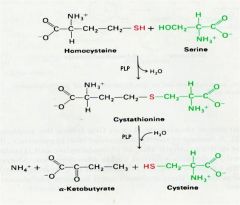
|
|
|
Methionine to homocysteine(SAM)?
SAM's fxn? |
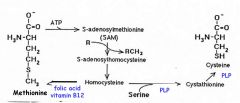
donor of methyl groups in acell, acceptors include DNA, protein, lipids, NTs
|
|
|
SAM:
how is methyl group of methionine activated? what to transfer of methyl to aceptors give rise to? donates methyl to? |
linkage to SAM
SAH norepinphrine-->epinephrine acetylserotonin-->melatonin phosphatidylethanolamine-->phosphatidycholline guanidinoacetate-->creatinine |
|
|
Regulation of methionine
regenerated to via? |
transfer of methyl from N5-methyltetrahydrofolate(-->tetrahydrofolate) to homocysteine
cofactors: B12, B6(pyridoxal phosphate), folic acid |
|
|
folic acid:
is reduced to? which plays a role in? |
-tetrahydrofolate
-carbon transfer rxns for purine and pyrimidine synthesis for AA metabolism |
|
|
Synthesis of thymidine role in cancer
REFERENCE |
-dihydrofolate reducatase[-- methotrexate+aminopterin(analogs of folate)
- FU --> Fdump irreversibly inhibits thymidylate synthase(suicide substrate) |
|
|
vitamin B12
synthesized? stored? what rxns? requires ___ made by ___ for ___ in ___ transport proteins that carry it to the liver are called? |
bacteria, stored in liver
1. homocysteine-->methionine(folate involved) 2. methylmalonylcoA-->succinylcoA(folate not involved) intrinsic factors(glycoprotein), gastric parietal cells, absorption, illeum transcobalamins |
|
|
Folate vs. Vitamin 12
both needed for? no B12 leads to? results in? reintroducing folate can? |
-Both need for conversion of homocytsteine to methionine
-folate trapped as N5 methyltetrahydydrofolate -megaoblastic anemia and neuropathy -correct anemia but not neuropathy |
|
|
folic Acid supplmentation
-deficiency of folic acid can cause what? -1998:-->? -too much folic acid? |
neural tube defects in pregnant women(spina bifida, anencephaly)
1998: mandate spplementation of grain products-->reduction in spina bifida 20% mask Vitamin B12 deficiency causing neurologic damage |
|
|
Amino Acid Catabolism:
direct sources of ammonia in liver? |
-NH3 neurotoxic is convered in liver to urea which is non-toxic and soluble-->excreted
-glutamate -gluatamine: transported form other tissues -alanine: transported from muscle for gluconeogensis and converted to pyruvate in liver genreating NH3 |
|
|
Urea cycle:
takes place? converts? enzymes located ? |
-liver
-converts NH3 to urea - 2 in MC, 3 in cytosol |
|
|
Urea cycle 3 facts
|
1. important
2. substrates are running around 3. Ornithine is essential and not consumed in the reaction |
|
|
Urea cycle regulation
|
1. transcription high when protein intake is high
2. Control of first enzyme; arginine activates N-acetyl glutamate synthetase allosterically |
|
|
Urea cycle malfxn
occurs when? most severe? mildest? also seen in? |
genetic disorders alter fxn of any of the enzymes
neonatal hyperammonemic coma, mental retardation, cerebral palsy episodic hyperammonemia precipitated by high protein intake/infection malfxn of urea cycle seen in liver failure(non-genetic) |
|
|
Malfxn of Urea cycle leads to?
2 hypothesis |
accumulation of ammonia in plasma and tissues(including brain) --> vomiting siezers, somnolence, coma and death
1. Toxicity of glutamine: high levels accumulate in astrocytes which have potent gluatmine synthesate leads to osmotic pressure(swelling and edema) 2. Depletion of a-ketoglutarates: high NH4-->a-ketoglut converted to gluatmate(via glut. dehydrogenase)-->decrease of TCA cycle-->decrease oxidative phosphorylation(neurons dependent) |
|
|
Managements of urea cycle defects
hyperammonemia --> treatments? long-term? |
protein catabolism
hemodialysis drug to conjugate glutamine -reduce dietary protein intake -provide adequate essential amino acids and non-protein calories -drugs to create alternate paths for N excretion |
|
|
Catabolism of carbon skeletons of amino acids
|
broken down into glycogenic/ketogenic products
glycogenic-->gluconeogeniss-->glucose ketogenic-->lipid metabolism -complicated pthways every step leaves possibility for mutation |
|
|
First step in degradation of phenylalanine
what is phenylketonuria? |
-Synthesis of tyrosine
* requires tetrahydrobipterin cofator, biopterin from GTP dihydrobiopterin + NADH --> tetrahydrobipterin + NAD *DHB reductatse deficiency of phenylalanine hydroxylase which prevents conversion of phenylalanine to tyrosine or deficiency of dihydrobipterin reductase(DHB reductase) or of biopterin synthesis |
|
|
Phenylkeotnuria management
|
dietary:
-diet low in phenylalanine -adequate other nutrients(tyrosine) -diet must be instituted very early; neonatal screening essential -diet must be maintained as long as possible Maternal PKU: -allows PKU women to reproduce -offspring had multiple birth defects -hyperphenylalanemia toxic to fetus -must have stringent control of blood phenylalanine during pregnancy |
|
|
Phenylketonura problems and solutions
|
cannot eliminate phenylalanine from diet,
avoid aspartame: aspartyl-phenylalanyl-methyl ester |
|
|
PKU II and other biopterin deficiencies
NTs affecteD? treated by? |
tetrahydrobiopterin(BH4) is a cofactor for reaction in pathways leading to NT synthesis:
tyrosine-->L-DOPA tryptophan--> 5-hydroxytryptophan(precursor of serotonin) maybe treated by providing BH4 |
|
|
Connecting glycolysis to lipogenesis
regulated by? |
Acetyl coA-->malonyl CoA
*acetyl CoA carboxylase -allosteric -hormonal: insulin -phosphorylated is inactive -dephosphorylated is active |
|
|
Fxns and Active forms of:
Glycogen synthase Glycogen Pase AcetylCoA carboxylase Pyruvate kinase PFK2(bifxnal) |
Glycogen synthase: deP
Glycogen Pase: P AcetylCoA carboxylase: deP Pyruvate kinase: deP PFK2(bifxnal): phosphatase domain active in P-form, kinase domain active in deP form |
|
|
Opposing actions of Insulin and glucagon/epi:
|
Insulin
+ glucose uptake, glycolysis(liver), glyocgen synthesis, fat synthesis - gluconeogenesis, glycogen breakdown, lipolysis, ketogenesis Glucagon/Epi - insulin +insulin |
|
|
Type 1 diabetes:
|
-No insulin
-glucagon unopposed -body keeps putting glucose into the blood, supply: -ammino acids-->gluconeogensis -FAs --> ketone bodies |
|
|
Type 2 diabetes:
|
-insulin present but doesn't match glucagon
-milder type 1 -no ketone bodies, though can get ketone bodies if severe enough(eventually looks like type 1) |
|
|
Alternative Fates of glucose in cell
|
G6P-->6 phosphogluconate-->ribose 5-P
also known as pentose phosphate pathway |
|
|
Pentose phosphate pathway
fxn? rate is controlled by? |
synthesis of pentose sugars for DNA, RNA, ATP, NADH FAD,
generate NADPH from NADP+ for biosynthetic rxns -NADP+ |
|
|
Roles of NADPH
|
1. Biosynthesis:
-FA -cholosterol -NTs -Nucelotides Reduced in Presence of NADPH 2. Detox 3. Reduction of oxidzed GSH erythrocytes: -keeps hemoglobin iron in ferrous state -stabilizes erythrocyte membrane |
|
|
1st step in pentose phosphate pathway:
need for? lack of leads to? effect on malaria? |
G6P-->6-phosphoglucono-d-lactone+NADPH+ H+ ---> 6 phosphogluconate
*G6Pate dehydrogenase *lactonase, H20 NADPH genereation hemolytic anema(heinz bodies), cells cannot maintained reduced gltuathione. protective against malaria |
|
|
Cancer cells convert pyruvate to lactate even in the presence of O2 why?
|
Need NADPH to make lipids
pentose intermediates to make DNA/RNA. trade energy loss go get more glucose into pentose phosphate shunt |