Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
231 Cards in this Set
- Front
- Back
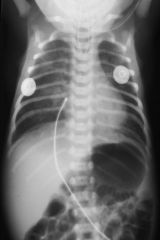
The following X-ray diagoses what condition in the neotate?
|
TE FISTULA
|
|
|
How frequently do infants with esophageal atresia have VACTERL association?
|
a third of the time
|
|
|
What can occur with maternal hypertension before the 20th week gestation?
|
Increased risk of miscarriage
|
|
|
What can occur with maternal hypertension after the 20th week gestation?
|
IUGR, placental insufficiency, placental abruption or previa.
|
|
|
What is associated with extensive hyperthermia for prolonged periods during days 14-30 post-conception?
|
Increased risk of neural tube defects.
|
|
|
What can occur with maternal infection with parvovirus B19?
|
congenital heart failure (hydrops) and death
|
|
|
What can occur with maternal infection with varicella?
|
limb reduction defects, chorioretinitis, skin scarring, developmental delay and microcephaly
|
|
|
What can occur due to maternal infection with rubella during the first 8 weeks gestation?
|
deafness (85%)
|
|
|
What can occur due to maternal infection with rubella during weeks 9-12 gestation?
|
cataracts (52%)
|
|
|
What can occur due to maternal infection with rubella during weeks 12-30 gestation?
|
heart defects (16%)
|
|
|
What can occur due to maternal infection with CMV during the first 27 weeks gestation?
|
low birth weight, mental retardation, microcephaly, periventricular calcifications and hearing loss that develops after the neonatal period
|
|
|
What can occur due to maternal infection with toxoplasmosis between weeks 10 and 24 of gestation?
|
hydrocephalus, blindness and mental retardation
|
|
|
What can occur due to maternal infection with syphillis after the first 5 months gestation?
|
abnormal teeth and bones, mental retardation and proteinuria
|
|
|
What are the three presentations of inborn errors of metabolism?
|
1) Intoxications
2) Energy defects 3) Complex molecule defects |
|
|
What should be suspected if a neonate or young infant suddenly presents with acute encephalopathy that is without warning and progresses rapidly.
|
Inborn errors of metabolism
|
|
|
What inborn errors of metabolism present with intoxication?
|
Amino acid diseases (maple syrup urine), hyperammonemias (OTC deficiency), organic acid diseases (popionic acidemia), sugar intolerances (galactosemia, fructose intolerance)
|
|
|
What inborn errors of metabolism present with energy disorders?
|
fatty acid oxidation defects, glycogen storage diseases and mitochondrial disorders
|
|
|
What inborn errors of metabolism present with complex molecule defects?
|
Hunter, Hurler, Albinism, Mevalonic aciduria, PDH deficiency, Zellweger syndrome, Infantile G-gangliosidosis, familial hypercholesterolemia
|
|
|
What is the most common feature of complex molecule defects?
|
hydrops
|
|
|
What are examples of complex molecule defects?
|
Lysosomal storage diseases, peroxisomal storage diseases, intracellular trafficking and processing defects and inborn errors of colesterol synthesis
|
|
|
How are peroxisomal disorders inherited?
|
All are AR except for X-linked adrenoleukodystrophy
|
|
|
How is hyperphenylalanimeia treated?
|
phenylalanine restriction, biopterin supplemetation and giving biogenic amine precursors (5-hydroxytryptophan and dopa)
|
|
|
How are tyrosinemia disorders inherited?
|
Autosomal Recessive
|
|
|
What is found in the urine in an infant with Type I tyrosinemia?
|
succinylacetone
|
|
|
How is PKU inherited?
|
Autosomal recessive
|
|
|
Patients with Tyrosinemia Type 1 have mental retardation. True or False?
|
False
|
|
|
Patients with Tyrosinemia Type 2 have mental retardation. True or False?
|
True
|
|
|
What presents in infancy as arrhythmias with severe cardiomyopathy and sudden death?
|
VLCAD
|
|
|
What is odor of sweaty feet associated with?
|
Isovaleric acidemia
and Glutaric Acidemia Type 2 |
|
|
What enzyme is defective in classic PKU?
|
Defect in Phenyalalnine Hydroxylase (PAH)
|
|
|
What IEM results from a defect in an enzyme carried on chromosome 12q24.1?
|
Classic PKU
enzyme is Phenylalanine Hydroxylase |
|
|
When does the damage from PKU become irreversible?
|
By age 8 weeks
|
|
|
What IEM has a "mousy" or "wolf-like" or "musty" odor?
|
Classic PKU
|
|
|
What illnesses are more common in Ashkenazi Jews?
|
Tay-Sachs 1/4000
Gaucher disease Type 1 (1/900) |
|
|
What presents as slowly progressive symptoms from buildup of toxic substances/metabolites that can include brain damage, loss of milestones, clots, strokes, etc?
|
Chronic Encephalopathy from IEM
|
|
|
What presents with sudden onset of seizures, coma, lethargy, hypertonia or hypotonia?
|
Acute Encephalopathy from IEM
|
|
|
What presents with fasting or illness that precipitates failure to thrive, hypotonia, cardiac dysfunction, lactic acidosis or weakness and fatigue?
|
Energy Defects in IEM
|
|
|
What group of disorders presents with severe progressive CNS dysfunction, facial dysmorphism, eye findings, renal cysts, hypotonia, abnormal MRI of brain, and hepatomegaly with liver dysfunction?
|
Peroxisomal Disorders
|
|
|
What is treatment of PKU?
|
Special diet FOR LIFE which includes phenylalanine
|
|
|
What affects occur when a mother who has PKU is not controlled during pregnancy?
|
"Maternal PKU"
-growth deficiency -microcephaly -mental retardation -congenital heart defects |
|
|
What should be suspected in someone who had elevated phenylalanine in blood, has been treated with dietary restrictions and now has normal phenylalanine levels in blood, but continues to have progressive neurologic problems?
|
Hyperphenylalaninemia
Defect in synthesis of tetrahydrobiopterin or defect in enzymes that regenerate biopterin from dihydrobiopterin |
|
|
What is clinically different between classic PKU and hyperphenylalanemia?
|
PKU progression stops with diet low in Phenylalanine.
HyperPhenylalanine progresses until phe is restricted AND BIOPTERIN, 5-hydroxytryptophan and dop are supplemented. |
|
|
How is classic PKU inherited?
|
Autosomal Recessive
|
|
|
How is tyrosinemia inherited?
|
Autosomal Recessive
|
|
|
What IEM can progress rapidly to death, but can also have a slower course of FTT, hepatomegaly, hepatoblastoma, renal tubular acidosis, Xray findings of rickets and NORMAL IQ?
|
Tyrosinemia Type 1
|
|
|
How is the liver failure and Fanconi Syndrome of hepato-renal tyrosinemia treated?
|
NTBC
2-(nitro-4-trifluoro-methyl-benzoyl)-1,3-cyclohexanedione And diet low in Phe and Tyrosine |
|
|
What is also known as Richner-Hanhart Syndrome?
|
Tyrosinemia Type II
or Oculocutaneous Tyrosinemia |
|
|
What IEM presents with corneal ulcers or dendritic keratosis, along with papular of keratotic lesions on palms and soles, with 50% of patients MR?
|
Tyrosinemia Type II
|
|
|
Are patients with Tyrosinemia Type I mentally retarded?
|
No
|
|
|
Are patients with Tyrosinemia Type II mentally retarded?
|
50% are
|
|
|
What enzyme is deficient in Alkaptonuria?
|
homogentisic acid dioxygenase
|
|
|
What IEM results from a deficiency of homogentisic acid dioxygenase?
|
Alkaptonuria
|
|
|
What disease is associated with "brick dust" in urine/diapers?
|
Alkaptonuria
|
|
|
What can present as severe Parkinsonism in infants?
|
AR deficiency of tyrosine hydroxylase (normal intelligence)
|
|
|
What IEM presents with symptoms at 3-5 days of life with rapid progression to death in 2-4 weeks without treatment? Symptoms: respiratory diff, feeding diff, loss of Moro reflex, seizures, etc.
|
Maple Syrup Urine Disease
|
|
|
What IEM has normal newborn with macrocephaly that decompensates with febrile illness leading to hypotonia, dystonia, subdural hematomas and retinal hemorrhages?
|
Glutaric Aciduria Type 1
|
|
|
What IEM can be mistaken for child abuse due to subdural hematomas and retinal hemorrhages?
|
Glutaric Aciduria Type 1
|
|
|
How do homocystinuria and Marfan's clinically differ?
|
Subluxation of lens is down in H and up in Mars.
H has limited joint mvmt and low IQ M is hyperextensible and nl IQ |
|
|
What vitamin can sometimes help treat homocystinuria?
|
Pyridoxine
|
|
|
What causes seizures in patients with nonketotic hyperglycinemia?
|
Accumulation of glycine in the CNS
|
|
|
Where is the highest concentration of glycine in nonketotic hyperglycinemia?
|
CNS
|
|
|
How is maple syrup urine disease diagnosed?
|
By finding increased Leucine, isoleucine and valine in the PLASMA and URINE
or finding ALLOISOLEUCINE |
|
|
Where does the urea cycle take place?
|
LIVER
|
|
|
What do the following IEMs share
-NAGS deficiency -CPS deficiency -OTC deficiency -Citrullinemia -Arginosuccinic aciduria -Arginemia |
They are urea cycle defects that cause hyperammonemia
|
|
|
How are urea cycle defects inherited?
|
All are AR except X-linked Ornithine transcarbamoylase deficiency (OTC)
|
|
|
What IEM presents in the first five days of life with respiratory alkalosis, mild or no liver dysfunction, no ketoacidosis and a LOW BUN?
|
Urea cycle defects
|
|
|
What is the treatment of urea cycle defects?
|
Restricting dietary nitrogen, replacing deficient amino acids, and pushing alternate pathways to eliminate nitrogen waste.
|
|
|
How is an infant with a urea cycle defect and a plasma ammonia level >200 micromols/L and/or COMA treated?
|
Hemodialysis
|
|
|
What IEM presents between ages 1 and 3 years with acute metabolic acidosis, hypoglycemia and carnitine deficiency during a stressor event?
|
3-methylcrotonyl-CoA carboxylase deficiency
|
|
|
How is 3-methylcrotonyl-CoA carboxylase deficiency (MCC) treated long-term?
|
Oral carnitine and biotin
|
|
|
What IEMs cause bone marrow suppression and pancreatitis?
|
Organic Acidemias
|
|
|
What IEMs produce the triad of alopecia, skin rash and encephalopathy?
|
Multiple Carboxylase Deficiency
|
|
|
What IEM presents early with hyperammonemia, ketoacidosis and thrombocytopenia or later with chronic ketotic hyperglycemia, vomiting, FTT & possible renal failure & cardiomyopathy?
|
Methylmalonic Acidemias
|
|
|
Where does fatty acid oxidation occur?
|
Mitochondria
|
|
|
What is the most common disorder affecting mitochondrial fatty acid oxidation and ketogenesis?
|
MCAD
medium-chain acyl-CoA dehydrogenase deficiency |
|
|
What presents in early infancy or later childhood with cardiomyopathy or recurrent episode of encephalopathy with hypoketotic hypogycemia? (also may have skeletal muscle weakness)
|
Primary Carnitine Deficiency
|
|
|
What IEM is associated with maternal acute fatty liver or HELLP syndrome?
|
Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHAD)
|
|
|
What IEM is frequently associated with development of cholestatic liver disease, retinopathy with hypopigmentation or focal pigment aggregations later in life?
|
LCHAD
|
|
|
What defect in fatty acid oxidation usually DOES NOT have arrythmias?
|
Carnitine Uptake Defect
Primary Carnitine Deficiency |
|
|
How is VLCAD (very long chain acyl-CoA dehydrogenase) deficiency diagnosed?
|
saturated and unsaturated C14-18 esters.
|
|
|
How are most glycogesn storage diseases inherited? What are the two exceptions?
|
Most are AR except 2 X-linked
phosphoglycerate kinase def and phosphorylase kinase def. |
|
|
What is the other name for lysosomal acid alpha-glucosidase deficiency?
|
Pompe's disease
GSD type 2 |
|
|
What is the other name for glucose-6-phosphatase deficiency?
|
Von Gierke's disease
GSD type 1 |
|
|
What IEM symptoms include feeding difficulties, irregular respirations, LOSS OF MORO REFLEX, seizures, opisthotonos & rigidity from cerebral edema?
|
Maple Syrup Urine Disease
|
|
|
What is the other name for Debrancher deficiency?
|
GSD type 3
|
|
|
What is the other name of liver phosphorylase kinase deficiency?
|
GSD type 9
|
|
|
What is the other name for myophosphorylase deficiency?
|
McArdle disease
GSD type 5 |
|
|
What glycogen storage disease is due to a defect in glucose-6-phosphatase in the liver, kidney and intestinal mucosa?
|
Von Gierke Disease
(type 1 GSD) |
|
|
When do patients with Von Gierke disease present?
|
3-4 months of age
|
|
|
How do patients with Von Gierke Disease present?
|
Hepatomegaly, failure to thrive and/or hypoglycemia with SEIZURES when infant starts to eat less often (3-4 months)
|
|
|
What IEM has doll-like faces with fat cheeks.
|
Von Gierke Disease
|
|
|
What liver effects occur in Von Gierke Disease?
|
Hepatomegaly with NORMAL liver transaminases.
|
|
|
What type of infections are more common in Von Gierke disease?
|
Type Ib has recurrent bacterial infections due to neutropenia and impaired neutrophil function.
|
|
|
How is Von Gierke Disease diagnosed?
|
Suspect with clinical finding abnormal lactate and lipid levels. Definitive diagnosis is gene-based mutation analysis.
|
|
|
What is treatment of Von Gierkes Disease?
|
Prevention of hypoglycemia - continuous NG feeds or oral uncooked cornstarch.
|
|
|
What are late effects of Von Gierke Disease?
|
By 20s-30s hepatic adenomas (sometimes malignant), pulmonary hypertension, osteoporosis, proteinuria, HTN, kidney stones and abnormal creatinine clearance, and focal segmental glomerulonephritis.
|
|
|
What medication is used in Von Gierke diesease to lower uric acid levels?
|
allopurinol
|
|
|
What ethnic group has an increased incidence of Type III GSD (Debrancher deficiency)?
|
Non-Ashkenazi Jews of North African descent.
|
|
|
What GSD presents with hepatomegaly, hypoglycemia, short stature, skeletal myopathy and/or cardiomyopathy?
|
Type III GSD
Debrancher Deficiency |
|
|
What percent of Type III GSD involves both liver and muscle abnormalities?
|
85%. The other 15% just involve the liver.
|
|
|
How can Type I and Type III GSD be differentiated clinically?
|
Type I liver and kidneys,
Type III kidneys are NOT enlarged. Type III, lactate and uric acid are normal. |
|
|
The most common presentation of which GSD is cirrhosis of the liver with hepatomegaly and FTT in the first 18 months of life with death occurring by age 5?
|
Type IV GSD
Branching Enzyme deficiency, amylopectinosis, Andersen Disease |
|
|
How does Type VI GSD affect a patient?
|
Mild hepatomegaly and growth retardation, but resolves by puberty. Benign course.
|
|
|
How do clinical effects of phosphorylase kinase deficiency differ if X-linked or AR.
|
X-linked is a mild disease with almost complete resolution by adulthood but small potential of cirrhosis.
AR is not benign. Many develop cirrhosis. |
|
|
What is the defect and result of Type 0 GSD?
|
glycogen synthase deficiency which leads to a decrease in glycogen stores.
|
|
|
Which GSD creates proximal renal tubular dysfunction and accumulation of glycogen in the liver and kidney.
|
Type XI GSD,
AKA Hepatic Glycogenesis with Renal Fanconi Syndrome, AKA Fanconi-Bickel Syndrome |
|
|
How do children with Fanconi-Bickel Syndrome present?
|
They present < 1 year of age with FTT, rickets and a large protuberant abdomen due to the hepatomegaly.
|
|
|
What type of GSD presents in children < 1 year of age with FTT, rickets and a large, protuberant abdomen due to the hepatomegaly.
|
GSD type XI
Fanconi-Bickel Syndrome |
|
|
What labs are abnornal in Fanconi-Bickel Syndrome?
|
Oral galactose and glucose-tolerance tests show impaired tolerance but liver transaminases are NORMAL despite significant hepatomegaly.
|
|
|
What GSD does not usually present until the patients are in their 20s or 30s and present with exercise induced muscle cramps and burgundy-colored urine after exercise?
|
McArdle Disease
(Type V GSD) |
|
|
What lab test is abnormal in patients with McArdle Disease?
|
Testing blood after exercise and finding increased ammonia instead of increased lactate.
|
|
|
What ethnic group has increased incidence of GSD type VII (Muscle phosphofructokinase deficiency)
|
Japanese descent or Ashkenazi Jews
|
|
|
With Type V GSD, what helps
|
glucose consumption BEFORE exercise
|
|
|
With Type VII GSD, what helps
|
avoiding carbohydrate loaded meals before exercise.
|
|
|
What GSD is aided by glucose consumption BEFORE exercise. Which is made worse by carbohydrates before exercise?
|
Type V glucose helps
Type VII glucose worsens. |
|
|
What is the defect in Pompe Disease?
|
a deficiency in the lysosomal acid alpha-1,4-glucosidase AKA acid maltase
|
|
|
Which of the age onsets with Pompe Disease is the most severe?
|
Infantile
|
|
|
How does infantile onset Pompe Disease present?
|
Cardiomegaly, hypotonia, and death before a year of age. Muscle weakness, macroglossia, hepatomegaly and hypertrophic cardiomyopathy.
|
|
|
How does juvenile/late childhood Pompe Disease present?
|
Slowly progressive skeletomuscular manifestations WITHOUT CARDIAC involvement.
|
|
|
How does adult form of Pompe Disease present?
|
Age 20-70s as a slowly progressive myopathy WITHOUT CARDIAC involvement. Presents with increased sleepiness, morning headache, and exertional dyspnea.
|
|
|
What labs are abnormal in Pompe Disease?
|
Elevated CPK, AST and LDH (especially in the infantile form)
|
|
|
What accumulates in patients with galactosemia?
|
Galactose-1-phosphate
|
|
|
What is the enzyme deficient in Galactosemia?
|
Galactose 1 phosphate uridyltransferase (GALT)
|
|
|
What test is used for Galactosemia screening in newborns?
|
Beutler test
|
|
|
What are long-term side effects of galactosemia?
|
-ovarian failure
-amenorrhea -developmental delay -learning disabilities that increase with age -speech disorders |
|
|
What is the presentation of fructokinase deficiency?
|
No clinical findiings, benign enzyme deficiency that requires no treatment.
|
|
|
What is a severe disease of infancy that occurs with ingestion of fructose-containing food?
|
Hereditary Fructose Intolerance (AKA deficiency of fructose 1,6-bisphosphate aldolase)
|
|
|
What presents like galactosemia but later in life? (Jaundice, hepatomegaly, vomiting, lethargy, seizures and irritability)
|
Hereditary Fructose Intolerance
|
|
|
What presents as severe episodes of metabolic acidosis, hypoglycemia, hyperventilation, seizures, and coma?
|
Fructose 1,6-diphosphatase deficiency
|
|
|
What is the presentation of fructokinase deficiency?
|
No clinical findiings, benign enzyme deficiency that requires no treatment.
|
|
|
What is a severe disease of infancy that occurs with ingestion of fructose-containing food?
|
Hereditary Fructose Intolerance (AKA deficiency of fructose 1,6-bisphosphate aldolase)
|
|
|
What presents like galactosemia but later in life?
(Jaundice, hepatomegaly, vomiting, lethargy, seizures and irritability) |
Hereditary Fructose Intolerance
|
|
|
What presents as severe episodes of metabolic acidosis, hypoglycemia, hyperventilation, seizures, and coma?
|
Fructose 1,6-diphosphatase deficiency
|
|
|
What is also known as MPS Type I?
|
Hurler Syndrome
|
|
|
What is also known as MPS Type II?
|
Hunter Syndrome
|
|
|
What is also known as MPS Type III?
|
Sanfilippo Syndrome
|
|
|
What is also known as MPS Type IV?
|
Morquio Syndrome
|
|
|
What presents in the first two years of life with coarsened facial features, mid-face hypoplasia and large tongues. They have frequent URIs and hernias.
|
Hurler Syndrome
|
|
|
What are patients with Hurler syndrome at high risk for?
|
Atlantoaxial subluxation
|
|
|
What MPS disease has corneal clouding?
|
Hurler Syndrome
|
|
|
What presents in the first two years of life with coarsened facial features, middle ear disease, learning difficulties, hernias, diarrhea, joint stiffness and HSM?
|
Hunter Syndrome
|
|
|
What is the only MPS disease that has X-linked inheritance?
|
Hunter Syndrome
|
|
|
Which has corneal clouding, Hunter's or Hurler's?
|
Hurler's
|
|
|
What is pathognomonic for Hunter Syndrome, but rare in children?
|
A nodular rash around the scapulae and the extensor surfaces.
|
|
|
What MPS can present with spondylolisthesis of L5/S1, and degenerative bone loss?
|
Hurler Syndrome
|
|
|
What can stem cell transplant do in Hurler Syndrome?
|
It CAN prevent intellectual deterioration and increase long-term survival. It CANNOT correct the skeletal abnormalities.
|
|
|
What MPS disease has dev del and URIs, then severe ADHD and aggression, then swallowing dysfunction and deterioration to vegetative state in the mid-teens and death by the 20s?
|
Sanfilliipo Syndrome
(MPS type III) |
|
|
What presents with short-trunk dwarfism, fine corneal deposits and skeletal dysplasia and normal intelligence.
|
Morguio Syndrome
MPS type IV |
|
|
What is the universal and most severe manifestation of Morquio Syndrome?
|
Odontoid dysplasia
|
|
|
What presents with a wide range of presentations from hydrops to juvenile sialidosis and a macular cherry-red spot?
|
ML I
(Cherry-red Spot Myoclonus Syndrome) |
|
|
What presents like Hurler syndrome with hyperplastic gums?
|
ML II
(I cell deficiency) |
|
|
What is life expectancy in ML I?
|
death occurs in late teens
|
|
|
What is life expectancy in ML II?
|
Death occurs early due to infection or cardiac failure.
|
|
|
What physical finding occurs in ML II but not in the other Mucolipidoses?
|
small head
|
|
|
What is life expectancy in ML III?
|
60s with 50% of patients with some LD or MR
|
|
|
What accumulates in sphingolipidoses?
|
ceramide
|
|
|
What are the 3 groups of sphingolipidoses?
|
1) globosides (red cell membranes and kidney)
2) gangliosides (gray matter in brain and terminals) 3) galactocerebrosides (cerebral white matter |
|
|
Infants with thick, shiny, collodion skin, HSM, hypertonic and hyperreflexive mvmts and poor suck?
|
Neonatal Gaucher
|
|
|
What presents with normal infant until 2-4 months when they have feeding difficulties and FTT, strabismus, opisthotonis and cherry-red macula?
|
Gaucher Disease
|
|
|
What presents with vomiting, diarrhea, FTT, HSM and cherry-red macular spots.
|
Niemann-Pick Disease Type A
|
|
|
What ethnic group is Niemann-Pick Disease Type A more common in?
|
Ashkenazi Jews
|
|
|
What has isolated, horizontal supranuclear gaze palsy?
|
Gaucher Disease Type 3
|
|
|
What presents in ages 3-5 with signs of ataxia and HSM and older kids with poor school performance and impaired fine motor skills?
|
Niemann-Pick Disease Type C
|
|
|
What sphinolipodoses has cataplexy and narcolepsy before death in the teen years?
|
Niemann-Pick Disease Type C
|
|
|
Verticle eye movements are lost and doll's eye movement preserved in this type of Niemann-Pick?
|
Type C
|
|
|
What presents in the first few months of life with exaggerated startle reflex that does not diminish with repeated stimuli and progressive loss of motor skills?
|
Tay-Sachs infantile form
|
|
|
How often do macular cherry-red spots
occur in the infantile form of Tay-Sachs Disease? |
90% of infants
|
|
|
What occurs in infantile Tay-Sachs with auditory stimuli?
|
seizures
|
|
|
What may present with clumsy, awkward gait, intention tremor, and dysarthria.
|
Juvenile/adult form of Tay-Sachs.
|
|
|
What is the most common lysosomal storage disease?
|
Gaucher Disease Type 1
|
|
|
What is the most common presentation of Gaucher Disease Type 1?
|
Splenomegaly
|
|
|
What is unusual about Gaucher disease type 1?
|
It has NO CNS involvement.
|
|
|
Which type of Gaucher Disease does not have CNS disease?
|
Type 1
|
|
|
Which type of Gaucher disease is the most common?
|
Type 1
|
|
|
What features are common to all forms fo Gaucher disease.
|
HSM, bone lesions and some lung disease
|
|
|
What is differene between type 2 and 3 Gaucher disease neurologic symptoms?
|
Type 3 neurologic disease is later and more chronic than type 2.
|
|
|
What percent of Gaucher mutations are in Ashkenazi Jews and how does this compare to non-Jewish population?
|
97% in Ashkenazi Jews
~75% in non-Jewish |
|
|
Niemann-Pick type B looks and presents clinically as what other storage disease?
|
Gaucher type 1
|
|
|
What is the only sphingolipidosis that is X-linked recessive.
|
Fabry disease
|
|
|
What has tiny, red-to-dark blue, papular lesions on the buttocks, scrotum, penis, buccal mucosa and umbilicus?
|
Angiokeratomata associated with Fabry disease
|
|
|
Urine showing Maltese crosses and casts are associated with what disease?
|
Fabry disease
|
|
|
What are Maltese crosses in urine?
|
birefringent lipid globules that are common in Fabry disease
|
|
|
What do the sphingolipidoses have in common?
|
They are all AR except the X-linked recessive Fabry disease, and are defects in the lysosoomal breakdown of sphingolipids causing buildup of ceramide.
|
|
|
Which types of Sphingolipidoses have CNS disease?
|
Tay-Sachs
Gaucher type 2 & 3 Niemann-Pick type A & C |
|
|
Which types of sphingolipidoses DO NOT have CNS disease?
|
Gaucher type 1
Niemann-Pick type B Fabry disease |
|
|
Which type of sphingolipidoses has only CNS disease?
|
Tay-Sachs
|
|
|
Which type of sphingolipidoses have CNS and HSM involvement?
|
Gaucherr type 2 & 3
Niemann-Pick type A & C |
|
|
Which type of sphingolipidoses have CNS with cardiac, renal, vascular or pulmonary involvement?
|
Niemann-Pick type A
|
|
|
Which type of sphingoliipidoses have predominantly HSM?
|
Gaucher type 1
Niemann-Pick type B |
|
|
Which type of sphingolipidoses have peripheral nervous system involvement?
(+/- skin, cardiac, renal, vascular or pulmonary) |
Fabry Disease
|
|
|
What are defined as organelles that have a single membrane, are filled with important enzymes that catabolize beta-oxidatioin of fatty acids, and are found in just about all cells except RBCs?
|
peroxisomes
|
|
|
What are the two classes of peroxisomal disorders?
|
1) peroxisomal biogenesis disorders which involve a deficiency of multiple peroxisome functions and
2) single-function disorders in which only one peroxisomal function is missing. |
|
|
What is characterized by high forehead, epicanthal folds, broad based nasal bridge, anteverted nares, micrognathia, large anterior fontanelle, cataracts and
pigmented retinopathy, hearing loss and vision loss? |
Zellweger Syndrome Spectrum. (severe end of peroxisome spectrum)
|
|
|
What peroxisomeal disorder presents between ages 3 and 10 with central demyelination and rapidly progresses in 1/3 to death.
|
X-linked adrenoleukodystrophy
|
|
|
When does X-linked adrenoleukodystrophy present?
|
age 3-10
|
|
|
When does adrenomyeloneurpathy present?
|
30s-40s
|
|
|
How does adrenomyeloneuropathy present?
|
gait disturbances, urinary sphincter dysfunction, adrenal insufficiency and cerebral effects.
|
|
|
What is the X-linked disease that results in impaired uptake of copper?
|
Menke's Disease
|
|
|
How is Menke's Disease inherited?
|
X-linked
|
|
|
What perinatal findings occur in Menke's Disease?
|
premature delivery,
temp instability, hypothermia, hypotonia and hypoglycemia |
|
|
What can Menke's Disease appear to be?
|
Shaken baby due to subdural hematomas and retinal hemorrhages.
|
|
|
What is the typical clinical course of Menke's Disease?
|
By 2-3 months, they are losing any milestones they've achieved, they have intracranial bleeds and death by age two.
|
|
|
What can have MR with any or all of the following: microcephaly, broad nasal tip, hypertelorism, cleft palate, micrognathia, anteverted nostils, ptosis, low-set ears, narrow bifrontal diameter, abnormal thumbs, congenital heart defects, polydactylty and/or syndactyly?
|
Smith-Lemli-Opitz
|
|
|
How is Smith-Lemli-Opitz inherited?
|
AR
|
|
|
Which types of porphyrias have neurologic abnormalities?
|
Hepatic porphyrias
|
|
|
Which types of porphyrias have cutaneous photosensitivity?
|
Eryhropoietic porphyrias
|
|
|
What are the most common drugs that can exaccerbate porphyria?
|
Barbiturates, Sulfonamide antibiotics, anti-seizure medications, griseofulvin and synthetic estrogens (BCPs)
|
|
|
What is the most common phenotypic expression of porphyria?
|
Asymptomatic heterozygotes until something trips the wire to increase production of pyrogens.
|
|
|
What is the most common symptom of acute intermittent porphyria?
|
Abdominal pain with ileus (Tenderness and fever are absent as this is neurologic and not inflammatory)
|
|
|
What test can be used to rule out acute intermittent pophyria in family members?
|
A normal PBG (porphobilinogen) in the stool.
|
|
|
Peripheral neurophahy due to axonal degeneration in porphyria affects which motor neurons preferentially?
|
The proximal muscles of the shoulders and arms.
|
|
|
What can result if acute porphyria is allowed to progress?
|
Progressive weakness can lead to respiratory and bulbar paralysis, seizures and mental symptoms of anxiety/depression and paranoia.
|
|
|
What is the most effective therapy for porphyria if given early in attack?
|
IV heme
|
|
|
Cutaneous porphyria has four types, but all have what in common as their primary defect?
|
They all have a deficiency of hepatic URO-decarboxylase.
|
|
|
What may precede or follow the bullae of porphyria in sun exposed areas?
|
milia
|
|
|
What excesses can contribute to the development of hepatic URO-decarboxylase deficiency?
|
Alcohol
Iron Estrogen (also hexachlorobenzene, dioxin, and chlorophenols) |
|
|
What type of cancer are patients with cutaneous porphyria at higher risk for?
|
Hepatocellular carcinoma
|
|
|
Of the cutaneous types of porphyria, which is sporadic?
|
Type 1
|
|
|
Of the cutaneous types of porphyria, which is a result of exposure?
|
Type 4
|
|
|
Of the cutaneous types of porphyria, which is familial?
|
Types 2 & 3 are familial
|
|
|
What type of porphyria also has hypertrichosis and hyperpigmentation?
|
Porphyria cutanea tarda
(cutaneous) |
|
|
What presents in infant boys as refractory hemolytic anemia, with pallor and weakness?
|
X-linked sideroblastic anemia due to deficient activity of erythroid form of aminolevulinate (ALA) synthase.
|
|
|
What vitamin may help treat porphyria?
|
B6 - pyridoxine
|
|
|
How is erythropoietic protoporphyria inherited?
|
EPP is an AD disorder.
|
|
|
How is the skin photosensitivity in EPP different from other porphyrias?
|
It does NOT have vesicles and pigment changes, severe scarring and hirsuitism are unusual.
|
|
|
How does EPP affect the liver?
|
Liver function usually normal, chronic liver disease is occasional, gallstones can occur and contain protoporphyrin.
|
|
|
What supplement can increase the tolerance to sunlight in porphyria?
|
Beta-carotene.
|
|
|
What is a rare X-linked disorder that causes gout, neurodevelopmental delay and sensorineural deafness?
|
Phosporibosyl pyrophosphate synthetase superactivity.
|
|
|
What is adenylate deaminase deficiency?
|
AR trait that presents as muscle weakness and cramping following vigorous exercise, but does not involve myoglobinuria or an abnormal muscle biopsy.
|
|
|
How is Lesch-Nyhan deficiency (HGPRT deficiency) inherited?
|
X-linked
|
|
|
What presents at 3-6 months with FTT, emesis and irritability and progresses to self mutilation, renal stones and gout?
|
Lesch-Nyhan Disease (HGPRT deficiency)
|