Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
24 Cards in this Set
- Front
- Back
- 3rd side (hint)
|
What are the classic originally described mitochondrial disorders?
|
-These are the classic originally described mitochondrial disorders
-described originally based on symptoms -Many of these will be discussed more in detail later!! 1. Alpers 2. Chronic progressive external ophthalmoplegia (CPEO) 3. Kearns-Sayre 4. Pearson 5. Leigh Syndrome 6. Neurologic weakness with Ataxia and Retinitis Pigmentosa -- NARP (like carp with an N) 7. Mitochondrial Encephalopathy, Lactic Acidosis, Stroke-like episodes -- MELAS (me-lass) 8. Myoclonic Epilepsy with Ragged Red Fibers -- MERRF (like nerf with an M) 9. Leber’s Hereditary Optic Neuropathy -- LHON |
|
|
|
CPEO
|
-Chronic progressive external ophthalmoplegia
-Progressive weakness or paralysis of the eye muscles |
|
|
|
Kearns-Sayre
|
Progressive external ophthalmoplegia (PEO), retinopathy, ataxia, heart block – conduction problems
|
|
|
|
Pearson
|
Bone marrow failure, exocrine pancreatic failure
|
|
|
|
MERRF
|
MERRF (like nerf with an m)
-Myoclonic Epilepsy with Ragged Red Fibers -Seizures and muscle disease |
|
|
|
LHON
|
-Leber’s Hereditary Optic Neuropathy
-Patients have a particular look to the retina |
|
|
|
What is MELAS?
When is the onset? How does it typically first present? Symptoms? |
MELAS (me-lass) = Mitochondrial Encephalopathy, Lactic Acidosis, Stroke-like episodes
-Kids with MELAS tend to spend a lot of time in the hospital -Childhood onset -->Typically toddlerhood but can be any age Symptoms: -Neurologic -->Typically first presents with non-specific neurologic symptoms -->Seizures, headaches, altered consciousness, transient clumsiness for a day or hemiparesis or blindness, almost like a MS picture where you get nonspecific neurologic symptoms that come and go -->Progressive impairment of motor, visual, and cognitive abilities -->As time goes on, more and more of these neurologic effects become permanent -Lactic acidosis -Myopathy -->Weakness, progressive hypotonia |
|
|
|
What are the genetics behind MELAS?
|
-mt-DNA mutations
80% in MT-TL1 (mitochondrial tRNA gene) -->1 common mutation (m.3243A>G) -->Will often check just for that mutation first because they can get the results really quickly Maternal inheritance – most cases -->Mom may have mild symptoms like migraines, headaches, short stature and her affected child will be much worse with full fledge MELAS Heteroplasmy and tissue distribution affects phenotype/degree of severity |
|
|
|
What is pyruvate dehydrogenase deficiency?
|
-PDH is the link between glycolysis and TCA (Krebs) cycle
-Multi-enzyme complex -->Multiple pieces of the complex are encoded for by different genes -->Multiple essential cofactors What are the cofactors??? |
-Thiamine
-α-lipoic acid -FAD, NAD, CoA |
|
|
How do kids with pyruvate dehydrogenase deficiency present?
|
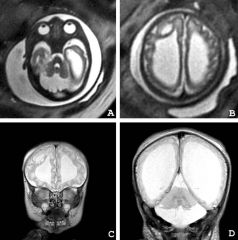
1. Psychomotor retardation
-->Intellectual disability 2. Hypotonia -->weakness 3. Progressive encephalopathy -->This can include Leigh syndrome 4. Brain malformations -->This depends on severity -->Brain development in utero is a very energetic process, so if you can’t do this well then you can have brain malformations at birth -->The brain malformations can be really bad -->There is NOT enough brain here -->The white is spinal fluid -->This little rim of brain here is what is left in these patients -->They do very poorly :( :( :( 5. Occasional extra-CNS manifestations -->Mostly neurologic disease, but can see some non-neurologic effects like: a. Cardiomyopathy b. Liver disease (These are pretty minor in PDH deficiency generally) |
|
|
|
What abnormality do the vast majority of patients with pyruvate dehydrogenase deficiency have ?
What type of inheritance pattern does PDH deficiency have? |
-Vast majority of patients with PDH deficiency have abnormalities of one particular subunit -- E1α subunit
-X-linked Why is the fact that it's X-linked interesting? |
-This is interesting because most genetically encoded metabolic diseases are recessive
-Males generally worse, though females may certainly be similarly severe depending on Lyonization X-inactivation patterns |
|
|
What is Leigh syndrome?
What else is it commonly called? When do patients present? |
Leigh Syndrome
-Neurologic degeneration, especially brainstem and cerebellum -->Progressive neurodegenerative disorder -Other aliases: Subacute necrotizing encephalomyelopathy -These kids are usually normal at birth and for the first few months of life -->Generally presents at 3-12 months of age -->Typically with mild infection (cold, flu, overnight fast) as trigger -Psychomotor regression – lose whatever skills they had -After the first hit, there is progressive neurologic degeneration – snowball effect -->Decompensation with each catabolic stress (Infection, fasting, etc). Stepwise loss of skills --> Hypotonia, spasticity, chorea (abnormal movements), ataxia, peripheral neuropathy -Cardiomyopathy *Most die by age 2-3 years |
|
|
|
How do we diagnose Leigh disease?
|
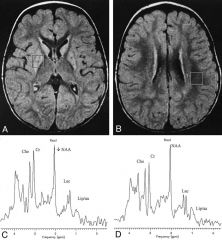
-Progressive neurologic disease with motor and intellectual delay
-Primarily affects the brainstem and/or basal ganglia disease -->A bit unusual in that there are a lot of neurologic degenerative diseases that affect the cortex, but not many that primarily affect the basal ganglia and brain stem -->If we see on neuroimaging a healthy looking cortex and a rotten looking brain stem and basal ganglia, we’re worried about Leigh syndrome -Elevated lactate in blood or CSF -One of: -->Typical LD features on neuroimaging a. Can have changes that are specific for Leigh disease -->Typical neuropathologic changes a. Multiple focal symmetric necrotic lesions b. Looking at brain under histopathology can give it away c. Typically need to have an autopsy for this --> Typical neuropathologic changes in affected sibling a. Family history __ -This is what Leigh syndrome looks like -This cortex looks pretty good, but what you see is kind of moth eaten basal ganglia -Note little punctate changes that aren’t supposed to be there -Can do spectroscopy on MRI and note high levels of __ acids (??? Min 30:45, can’t understand him) here, lactates here, you shouldn’t see a peak of lactate there |
|
|
|
What causes Leigh syndrome?
|
-It’s complicated
-There are many different genes that can be affected – most can be either mitochondrial or nuclear genes that lead to the same phenotype -Mostly oxidative phosphorylative function. -In figuring out which one it is is really important for the families for recurrence risk counseling Here's the list: Complex I genes -->mitochondrial-encoded MTND3 , MTND5, and MTND6 -->nuclear-encoded NDUFV1, NDUFS1, NDUFS3, NDUFS4, NDUFS7, and NDUFS8 Complex II gene: -->the flavoprotein subunit A (SDHA) Complex III gene: -->BCS1L which is involved in the assembly of complex III Complex IV genes -->mitochondrial-encoded MTCO3 nuclear-encoded COX10, COX15, SCO2, and SURF1 -->SURF1 is involved in the assembly of complex IV. Complex V gene: -->the mitochondrial-encoded MTATP6 This is a list from ~1 year ago, but they keep adding new ones |
|
|
|
How is Leigh syndrome an example of a genotype to phenotype correlation?
Name two genetic changes that occur, and we'll go from there! |
1. T8993G
2. T8993C What do patients present with for each mutation if their mutant load is: <60%? ~70-90%? >90%? |
1. T8993G
-->Mutant load <60% assymptomatic -you’re fine – no symptoms, no way of knowing you have it -->Mutant load ~70-90% have NARP -NARP (like carp with an n) -Neurologic weakness with Ataxia and Retinitis Pigmentosa -Late-onset peripheral neuropathy, ataxia and RP -Also retinal degeneration -Patients with NARP typically live into adulthood, they may not live well but it’s very different than Leigh -Peripheral neuropathy, ataxia, retinal degeneration -->Mutant load >90% have Leigh syndrome *** This is a typical classic example of heteroplasmy effects*** 2. T8993C -->Less severe -->Mutant load <90% assymptomatic -->For patients to have symptoms, patients need >90% mitochondria with the T8893C mutation Can display as full fledged Leigh syndrome if you have the right/wrong heteroplasmy **Can also have GENOTYPE effects A different mutation at the same locus – in this case, instead of mutating T8893 to G, it’s T8893C -This has a less severe phenotype |
|
|
What is NARP?
|
-If ~70-90% of your mitochondria have the T8893G mutation, you have NARP
-NARP (like carp with an n) -Neurologic weakness with Ataxia and Retinitis Pigmentosa -Late-onset peripheral neuropathy, ataxia and RP -Also retinal degeneration -Patients with NARP typically live into adulthood, they may not live well but it’s very different than Leigh |
|
|
|
What are POLG1 related disorders?
Also, what even is POLG1? What does it do? How does it affect the mitochondria? |
POLG1 (DNA polymerase subunit gamma) – related disorders
-Nuclear gene -->Present on chromosome 15 -Plays a role in mitochondrial DNA maintenance and replication -3 main functional domains (some genotype/phenotype correlation) 1. Polymerase 2. Exonuclease 3. Linker -How this affects mitochondria is by causing depletion of DNA within mitochondria -->Reduction in copy number of mtDNA -->Depletion is also caused by other genes and result in similar phenotypes with either brain, liver, or both diseases, although polymerase gamma is by far the most common cause of DNA depletion -->DGOUK, MPV17, TK2, others -Have normal numbers of mitochondria, but they progressively lose their genome and as they lose their genome they wont make substrates for the respiratory chain or the tRNAs they need and you’ll have progressive mitochondrial dysfunction -Continuum of overlapping phenotypes -->Generally cause either hepatocerebral disease (like POLG1) or encephalomyopathic disease -->Because symptoms are so vague and can be at any different age, they check of this a lot -->He’s found it in a couple different families, but they check for it more than they find it -Range in severity, organ specificity, and age of onset -All originally described separately and nobody realized they had a common cause until the molecular genetic techniques caught up to the clinical descriptions of the diseases -May be identified based on direct mutation detection or by mtDNA depletion -No specific effective therapy -Not much to offer in terms of cures or therapy |
|
|
|
What is Alpers?
What causes it? Symptoms? What's the first symptom typically? Age of onset? |
SEIZURES – NEUROLOGIC DEGENRATION – LIVER DYSFUNCTION
-Also caused by POLG (DNA polymerase subunit gamma) mutations Alpers -->Hypotonia (poor tone), seizures, liver failure Severe and progressive encephalopathy -Brain dysfunction – have seizures, severe intellectual disability -Neuroimaging with gliosis (scar-like appearance), atrophy -Cortex is much more involved than the brainstem and basal ganglia (opposite of Leigh) -->Sometimes will have a free floating brain stem and basal ganglia and the cortex is mostly gone with this disease -Seizures often first symptom -->That can happen at any age -->He’s had patients that seemed fine, and then at 10 months they had a seizure and by 2 years they were dead :( meep. -Can progress very quickly -Age of onset variable (2-4y typical) -->Most common descriptions are in toddlerhood but he’s seen babies and older kids -Liver dysfunction What precipitates the liver dysfunction? |
->May be precipitated by anticonvulsants
some anti-seizure medications are clearly responsible for the liver dysfunction associated with this, but other kids will have liver dysfunction without having been exposed to these anti-seizure medications -Variable progression to liver failure -Can be minor to sever liver disease -Can progress to full on liver failure and cirrhosis -Liver disease itself usually does not kill these patients, its mostly the neurologic problems |
|
|
What are diseases caused by POLG1 (DNA polymerase subunit gamma)?
|
1. Alpers
2. Childhood Myocerebrohepatopathy 3. Myoclonic epilepsy myopathy sensory ataxia (MEMSA) --"Meem-sah” 4. Ataxia neuropathy spectrum 5. AR and AD PEO (Autosomal recessive/dominant progressive external ophthalmoplegia) -- eye movement problems |
|
|
|
What is Childhood Myocerebrohepatopathy?
|
-Similar to Alpers in a lot of ways but it’s milder and has later onset
-Seizures, neurologic regression -Liver dysfunction -->Less of a cirrhotic liver dysfunction and more of a synthetic liver disease – not making coagulation factors and things like that |
|
|
|
What is Ataxia neuropathy spectrum (ANS)?
|
Children with more movement disorders and peripheral neuropathy can be caused by POLG1
|
|
|
|
What happens if you have just one POLG1 mutation?
|
-If you have just one POLG1 mutation (carrier) then you can have doubled vision, eye movement issues
-All of these are more severe phenotypes that are caused by recessive disease (having mutations on both copies of your polymerase gamma gene) |
|
|
|
What is MNGIE?
|
-Mitochondrial NeuroGastroIntestinal Encephalopathy
-Symptoms start in adolescence (variable) 1. Usually a gastrointestinal disease first. Gastrointestinal symptoms: a. Severe GI dysmotility -->Intestines simply won’t move. It’s like they have a post surgical illeus their whole life b. Cachexia (flip for more on cachexia) c. Nausea, vomiting, constipation, incontinence (sphincter dysfunction) -->Pretty socially debilitating early on -Can be diagnosed with anorexia early on because they’ll be so skinny and so uncomfortable when they eat they just won’t, but it’s really a different phenomenon than anorexia 2. Peripheral neuropathy 3. Opthalmoplegia/Ptosis (flip for more on this!) |
Cachexia ( from Greek kakos "bad" and hexis "condition“ – this was too funny not to include)[1] or wasting syndrome is loss of weight, muscle atrophy, fatigue, weakness, and significant loss of appetite in someone who is not actively trying to lose weight. The formal definition of cachexia is the loss of body mass that cannot be reversed nutritionally: Even if the affected patient eats more calories, lean body mass will be lost, indicating a primary pathology is in place.
Opthalmoplegia: paralysis of one or more extraocular muscles which are responsible for eye movements Ptosis = drooping of the eyelids |
|
|
What are the genetics behind MNGIE?
|
-Caused by Thymidine phosphorylase deficiency
-->Nuclear encoded, cytoplasmic protein that causes a mitochondrial disease -->he’s literally having a braingasm over this, you can hear it in his voice **Autosomal recessive -People with thymidine phosphorylase deficiency have massive elevations of thymidine in their cytoplasm and mitochondria and in their blood stream == looking for high levels of thymidine is the main way to diagnose this disease -->Thymidine phosphorylase is present in the cytoplasm and helps to break down thymidine -Having high levels of thymidine levels around disrupts mitochondrial DNA replication system -->A lot of the error prevention part of the mitochondrial DNA duplication comes from tight control of nucleotide availability -->When you have this crazy amount of high thymidine around it introduces new mutations into the mitochondria as time goes on -->Leads to progressive accumulation of various mt-DNA mutations |
|