Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
23 Cards in this Set
- Front
- Back
|
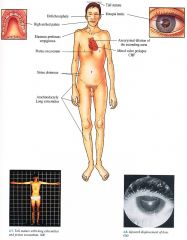
Clinical Marfan Syndrome
|
|
|
|
Inheritance
|
Autosomal dominant; mutation in fibrillin 1 on chromosome 15
|
|
|
Prenatal
|
DNA analysis
|
|
|
Incidence
|
1: 10 000 2 0, 000; M = F
|
|
|
Age at Presentation
|
Infancy if suspected by family history; usually second or third decade of life
|
|
|
Pathogenesis
|
Mutation in fibrillin gene, coding for a vital component of the microfibrillar system. results in a lack of fibrillin with concomitant defects in the ocular, cardiovascular, ane musculoskeletal system
|
|
|
Clinical
|
Musculoskeletal
TalI stature, lower body length longer than upper body length, arachnoclactyly, dolichocephaly, pectus excavatum, high arched palate, loose joints, poor muscle tone, kyphoscoliosis, pes planus, inguinal hernia Eyes Ectopia lentis (upward displacement in 75%) Myopia Cardiovascular Progressive aneurysmal dilatation of ascending aorta with secondary regurgitatior (CHF), dissection and rupture Mitral valve prolapsed Skin (less common) Striae distensae Elastosis perforans serpiginosa Decreased SQ fat |
|
|
D/Dx
|
Congenital contractural arachnoclactyly; Multiple endocrine neoplasia type Ilb (p. 190); Homocystinuria (p. 332) ;Stickler syndrome
|
|
|
Lab
|
Echocardiagram Chest x ray
|
|
|
Management
|
Referral to cardiologist/cardiac Surgeon surgical repair, B blockers Referral to ophthalmologist Referral to orthopedic surgeon
Estrogen therapy prevent excessive tallness in females |
|
|
Prognosis
|
Although the prognosis has improved dramatically with advanced cardiovascular surgical repair, patients may die prematurely from cardiac complications; marked variability in severity
|
|
|
Clinical Cutis Laxa
|

|
|
|
Synonym
|
Generalized Elastolysis
|
|
|
Inheritance
|
Autosomal recessive (most common) FBLN5 (fibulin 5) gene on 14q32; another locus on 5q23 31;
Autosomal dominant elastin gene on 7ql I and FBLN5 on 14q32 (usually skin only); X linked recessive ATP7A on Xq12 13; acquired |
|
|
Prenatal
|
DNA mutation analysis
|
|
|
Incidence
|
Rare
|
|
|
Age at Presentation
|
Birth to infancy
|
|
|
Pathogenesis
|
Heterogeneous mutations in fibulin 5 gene, elastin gene, or ATP7A gene (adenosine triphosphatase [ATPasel mutation that impairs copper transport necessary for lysyl oxidase. activity and normal elastin production) contributes to variability in clinical severity
|
|
|
Clinical
|
Skin
Loose, redundant, pendulous skin folds with hound dog facies, often generalized: elastic, lacks recoil Premature aged appearance Oral Vocal cord laxity causing deep, resonant voice Lungs Newborn hypoplastic lungs Emphysema (autosomal recessive) may be complicated by tachypnea, pneumontia, cor pulmonale Gastrointestinal (autosornal recessive) Esophageal, duodenal, rectal diverticulae Genitourinary (autosomal recessive and x linked) Bladder diverticulae Musculoskeletal (autosornal recessive and X linked) Inguinal, diaphragmatic, umbilical hernia, hip dislocation, occipital horn exostoses (x linked) |
|
|
D/Dx
|
Pseudoxanthorna elasticum (p. 144) EDS (p. 134) Granulomatous slack skin DeBarsy syndrome SCARF syndrome
|
|
|
Lab
|
Skin biopsy decreased, fragmented elastic fibers visualized with Verhoeff van Giesenstain;
Serum copper and ceruloplasmin levels Chest x ray |
|
|
Management
|
Referral to plastic surgeon Referral to pulmonologist, gastroenterologist, urologist, surgeon if symptomatic Sunscreen protection
|
|
|
Prognosis
|
Great variability in severity ranging from death in neonate if born with hypoplastic lung
to only skin involvement with normal life span if no pulmonary disease (majority with latter. |