Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
23 Cards in this Set
- Front
- Back
|
What are the basics of drug actions?
|
drugs function by interacting with a cellular 'target'
eg, protein, DNA, RNA, lipids many targets are enzymes or transporters (eg, inhibitors) or receptors (eg, agonists and antagonists) |
|
|
How do drugs have selective toxicicy?
|
unique target in parasite
- chloroquine blocks hemazoin formation discrimination between host and parasite targets - pyrimethamine and DHFR target is more important to parasite than host - DNA synthesis (antifolates) drug activation by parasite - anaerobes and nitroimidazoles greater drug accumulation by parasite - food vacuole and chloroquine |
|
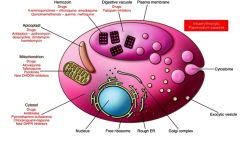
What are the major drug targets?
|
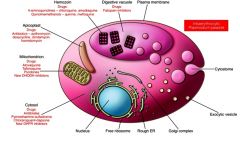
food vacuole and digestion of hemoglobin.
As the parasite matures, it develops a special organelle, called the cytostome, for the uptake of host cytoplasm. The hemoglobin-containing vesicles fuse to form a food vacuole. The food vacuole is an acidic compartment (pH 5.0–5.4) that contains protease activities, and in this regard resembles a lysosome. Digestion of hemoglobin also releases heme. Free heme is toxic due to its ability to destabilize and lyse membranes, as well as inhibit the activity of several enzymes. Some of this free heme can be degraded in oxidative processes. However, the majority of the heme is sequestered into hemozoin or the malarial pigment |
|
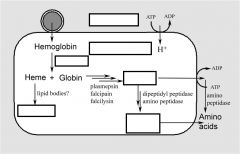
How and where is hemoglobin digested?
Identify unlabeled parts. |
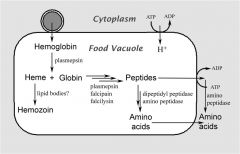
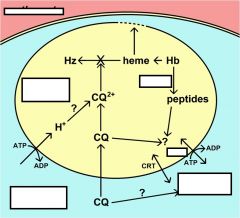
Hemoglobinvis taken up by the parasite via pinocytosis and delivered to the food vacuole where proteases break down the globin into peptides and amino acids that are transported to the host erythrocyte. The heme is crystallized into hemozoin resulting in its detoxification.
The malaria parasite takes up the host erythrocyte cytoplasm and breaks down the hemoglobin into amino acids. These amino acids are then used for the synthesis of parasite proteins or possibly as an energy source. The food vacuole is an acidic compartment (pH 5.0–5.4) that contains protease activities, and in this regard resembles a lysosome. Aspartic acid proteases designated as plasmepsins make the initial cleavages of the native hemoglobin causing it to dissociate into large globin fragments and free heme. The globin fragments are then further digested by plasmepsins and falcipains (cysteine proteases) into peptides. Digestion of hemoglobin also releases heme. Free heme is toxic due to its ability to destabilize and lyse membranes, as well as inhibit the activity of several enzymes. Some of this free heme can be degraded in oxidative processes. However, the majority of the heme is sequestered into hemozoin or the malarial pigment |
|
How is chloroquine metabolized?
How does this effect plasmodium? |
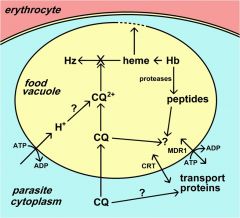
Chloroquine and other 4-aminoquinolines inhibit pigment formation, as well as the heme degradative processes, and thereby prevent the detoxification of heme.
The free heme destabilizes the food vacuolar membrane and other membranes and leads to the death of the parasite. The fact that the biocrystallization of heme is a unique process to the parasite and not found in the host accounts for the high therapeutic index of such drugs in the absence of drug resistance. |
|
Describe antifolates and nucleotide metabolism
|
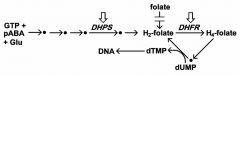
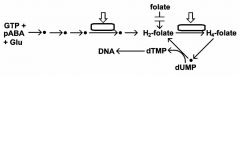
folate is necessary co-factor for nucleotide (i.e., DNA) synthesis and other biosynthesis
pyrimethamine preferentially inhibits parasite DHFR, dihydrofolate reductase sulfadoxine inhibits folate synthesis dihydropteroate synthetase(DHPS) - parasite cannot salvage folates - host only salvages |
|

What are the drug resistance levels?
|

Traditionally these levels of drug resistance have been defined as sensitive (no recrudescence), RI (delayed recrudescence), RII (early recrudescence), and RIII (minimal or no antiparasite effect). Drug resistance by this protocol is determined by monitoring patients for parasitemia for 28 days following standard drug treatment.
Because of the difficulties in using this protocol in endemic areas a modified protocol based on clinical outcome was introduced by the World Health Organization |
|
|
What is the definition of drug resistance?
|
Drug resistance is defined by a treatment failure and can be graded into different levels depending on the timing of the recrudescence following treatment.
parasite tolerance of higher drug levels |
|
|
What factors do you have to rule out before saying a drug is resistant?
|
non-compliance
bad quality wrong dose vomiting |
|
|
What is the modified drug resistance protocol?
|
introduced by WHO in 1996
based on clinical outcome: - adequate clinical response (ACR) - late treatment failure (LTF) - early treatment failure (ETF) more practical in areas of intense transmission - difficult to distinguish re-infection from recrudescense - parasitemia in the absence of clinical symptoms is common |
|
|
What is the definition of adequate clinical response (ACR)?
|
no recrudescence by day 14
ACR is defined as the absence of parasitemia (irrespective of fever) or absence of clinical symptoms (irrespective of parasitemia) during 14 days following treatment. |
|
|
What is the definition of late treatment failure (LTF)?
|
recrudescence during days 4-14
LTF is the reappearance of symptoms or the presence of parasitemia during days four to 14 of follow-up. |
|
|
What is the definition of early treatment failure (ETF)?
|
persistence during days 1-3
ETF is the persistence of clinical symptoms or the presence of parasitemia during the first three days of follow-up. |
|
|
What are the drug resistance mechanisms?
|
mutations in target gene
increase production of target (gene amplification) drug inactivation decrease drug accumulation (includes increase efflux) |
|
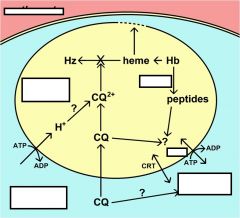
What is Multiple Drug Resistance (MDR)?
|
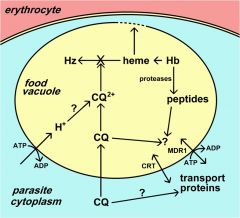
originally identified in drug resistant bacteria and cancer
ubiquitous family of ABC transporters (aka P-glycoprotein) Chloroquine resistance is associated with a decrease in the amount of chloroquine that accumulates in the food vacuole. The mechanism for this decreased accumulation is controversial. However, some studies have shown that the decrease in drug accumulation is due to an increase in drug efflux. The observation that verapamil and related drugs can reverse the chloroquine resistant phenotype has led to speculation that an ATP-dependent transporter plays a role in drug efflux and chloroquine resistance, similar to the multidrug resistance (MDR) in cancer. An MDR-like transporter, designated PfMDR1, has been identified on the food vacuole membrane. However, no definitive correlations between PfMDR1 and chloroquine resistance could be demonstrated. An ancillary role for PfMDR1 in chloroquine resistance cannot be ruled out though. |
|
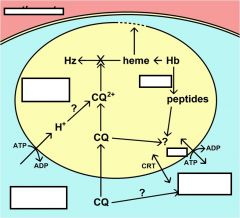
what is chloroquine resistance trasnporter (CRT)?
|
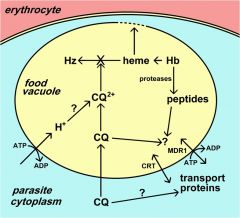
originally identified in cross between sensitive and resistance strains (saw and increase in polymorphisms)
A genetic cross and mapping studies between a chloroquine resistant clone and a chloroquine sensitive clone resulted in the identification of a 36 kb region on chromosome 7 associated with chloroquine resistance. The gene has been designated as PfCRT (for P. falciparum chloroquine resistance transporter) and the protein is localized to the food vacuole membrane. Several mutations in the PfCRT gene show correlations with the chloroquine resistance phenotype and one mutation, a substitution of a threonine (T) for a lysine (K) at residue 76 (K76T) shows good correlation with chloroquine resistance. Presumably these mutations affect the accumulation of chloroquine in the food vacuole, but the exact mechanism of chloroquine resistance is not known. |
|
|
What is the importance of point mutations in drug resistance?
|
point mutations seem to predominate as mechanism of drug resistance
selectable trait under drug pressure not necessarily a biological advantage molecular markers for evaluating drug resistance in populations rapid in vitro test for resistance (eg, PCR, gene chips) |
|
|
What is the major polymorphism for CRT, chloroquine resistance transporter?
- Name its subcellular location, primary function, and major drugs affected. |
Major Polymorphism: K76T
Subcellular location: food vacuole Primary function: transporter Major Drugs Affected: chloroquine |
|
|
What is the major polymorphism forMDR1, multidrug resistance (P-glycoprotein homolog)?
- Name its subcellular location, primary function, and major drugs affected. |
Major Polymorphism: Amplification, D86Y
Subcellular location: food vacuole Primary function: putative transporter Major Drugs Affected: Mefloquine, quinine (?) |
|
|
For protein, DHFR, dihydrofolate
reductase - - Name its subcellular location, primary function, and major drugs affected. |
Subcellular location:cytoplasm
Primary function: Folate metabolism Major Drugs Affected: Pyrimethamine, proguanil |
|
|
For protein, DHPS, dihydropteroate synthetase -
- Name its subcellular location, primary function, and major drugs affected. |
Subcellular location:cytoplasm
Primary function: Folate metabolism Major Drugs Affected: Sulfadoxine, dapsone |
|
|
How does drug resistance deveolop?
|
Drug resistance develops when parasites with decreased sensitivities
to antimalarial drugs are selected under drug pressure. Decreased drug sensitivity can be conferred by several mechanisms and reflects genetic mutations or polymorphisms in the parasite population. The drug-resistant parasites will have a selective advantage over the drug-sensitive parasites in the presence of drug and will be preferentially transmitted. Major factors in the development of drug resistance are the use of subtherapeutic doses of drugs or not completing the treatment regimen. |
|
|
What are the factors that contribute to developing and spread of drug resistance?
|
Self-treatment - Individuals may only take the drug until symptoms clear or will take lower doses to save money
Poor compliance - Individuals may not complete the full course of treatment because of drug side effects Mass administration - The widespread use of a drug in an area of intense transmission increases drug pressure by exposing a larger parasite population to the drug Long drug half-life - Drugs that are slowly eliminated will lead to a longer exposure of the parasite to subtherapeutic drug concentrations Transmission intensity - High levels of transmission may allow reinfection while drugs are at subtherapeutic levels |