Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
145 Cards in this Set
- Front
- Back
|
What enzyme does high acetyl CoA inhibit? What does it stimulate?
|
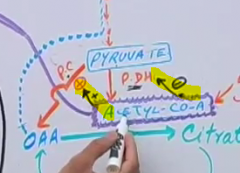
|
|
|
What happens to acetyl CoA when OAA is being dierted to gluconeogenesis?
|
it becomes used for ketogenesis
|
|
|
What organelle and organ does ketogenesis take place in>
|
the liver mitochondria
|
|
|
What are the ketone bodies?
|
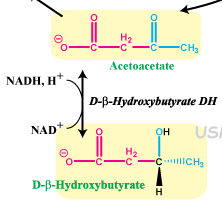
the ones that float in the blood
acetoacetate and b-hydroxybutyrate |
|
|
In your own words, narrate what happens in ketogenesis.
show reaction |
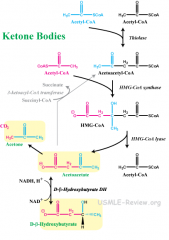
Liver mitochondria- you have lots of acetylCoA lying around so it joins up with acetoacetyl CoA, which is like the last step of beta oxidation before the last two acetyls break apart except the acetyl CoA are fused together. They join to make HMG CoA, but split again to make acetoacetate and regenerate acetyl CoA (but different carbons).
Acetoacetate can either be reduced to B-hydroxybutyrate or lose a CO2 and become acetone. Both travel in the blood. Acetoacetate and B-hydroxybutyrate are the ketones, both with oxidized B carbons. WHen they enter cells, the invested reduction of b-hydroxybutyrate is taken away and it all become acetoacetate. CoA's are taken from succinylCoA and slapped onto acetoacetate to cleave it into two acetyl CoA's to be fed into the Kreb's cycle. |
|
|
In a nutshell, what is transferred where in ketogenesis?
|
2 acetyl CoA are joined together and possibly given an extra reduction (NADH=3ATP) and then transferred from liver mitochondria to other cells for final cleavage into 2 acetyl CoA and Krebs energy
|
|
|
What determines if a tissue can use ketones?
|
if it has the enzyme to transfer CoA from succinyl CoA to acetoacetic acid.
(thiotransferase) |
|
|
What tissues can use ketones and under what conditions?
|
normal- muscle, kidney
in starvation- brain |
|
|
How is acetone production regulated? Where is it made?
Why do we get the alcohol smell? |
it happens spontaneously in the liver mitochondria from acetoacetate decarboxylation
high amounts of ketogenesis will produce it as a side product |
|
|
Under what blood conditions does ketogenesis happen? Why does this make sense?
|
high FA's in blood and low glucose.
you use the end products of b-oxidation so they better be plentiful! |
|
|
In even more of a nutshell, what is happening in ketogenesis?
|
the liver is chewing up FA's for the rest of the body (muscle, kidney, and brain)
|
|
|
How long do you need to fast to be in starvation for the brain to use ketones?
|
3-5 days
|
|
|
What other process is happening at the same time as ketogenesis?
Why does this balance out? |
GLUCOneogenesis
OAA is used for gluconeogenesis Unincorporated Acetyl CoA used for ketogenesis |
|
|
Why is the brain only able to use ketones in starvation?
|
level reach high enough to diffuse there
|
|
|
How many ATP are used in ketone utilization? From where?
|
1 ATP to transfer CoA to ketone
|
|
|
How many ATP does acetoacetate vs b-hydroxybutyrate yield?
|
acetoacetate- 23
b-hydroxybutyrate- 26 |
|
|
How would you test for ketones in blood?
|
look for it in the urine
|
|
|
What kind of deficiency would cause macro vs microcytic anemia?
|
macro- B12 and folate
micro- iron |
|
|
Why, generally, does diabetes II cause weight gain?
|
it promotes anabolism rather then catabolism
|
|
|
What is the point of the urea cycle?
|
to take NH4+ left over from metabolizing AA's and transform them into urea to be excreted
|
|
|
Where does urea cycle happen?
organ and organelles? |
in the liver mitochondria (1st) and cytoplasm
|
|
|
WHat happens in the mitochondria vs the cytoplasm?
|
mitochondria- NH4+ left over from AA metabolism is collected and secured into new compound
cytoplasm- urea is actually made in a cycle and excreted to blood |
|
|
Where do the 2 NH2 in urea come from?
|
1 from AA's metabolized
1 donated from aspartate |
|
|
What does aspartate become when it's NH3 is taken away like that?
|
part of a intermediate where it is essentially succinate
|
|
|
Where and what is the rate limiting step for the urea cycle?
Why is this a good control point? |
Carbamoyl phosphate synthase in the liver mitochondria
You only want to stimulate thew urea cycle for as much as you need to dump nitrogen made in the mitochondria |
|
|
What is a great mnemonic to remember the first and last intermediate in urea cycle?
|
they begin with the same letters as the TCA cycle!
Ornithine Citrulline |
|
|
Mnemonic for aspartate doing the NH3 donation?
|
ass is really close to urethra
ASSpartate! |
|
|
What does carbamoyl phosphate look like?
|
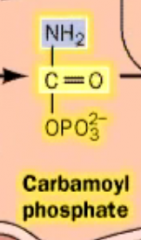
|
|
|
Is it free to make carbamoyl phosphate?
|
no it costs 2 ATP
|
|
|
What is the actual urea cleaved off of?
|
arginine
|
|
|
show arginine. How many CH2 does it have till the C(NH3)3?
Mnemonic? |
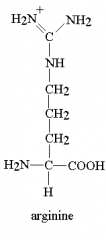
Arginine has 3 NH3 and 3 CH2
|
|
|
Imagine the structural changes within the urea cycle
|
|
|
name these intermediates
|
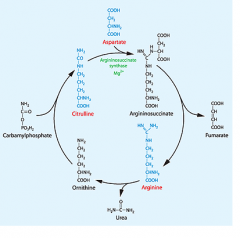
|
|
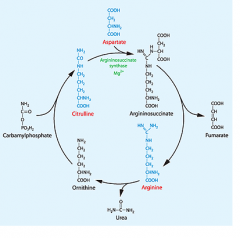
highlight where the urea comes from
|
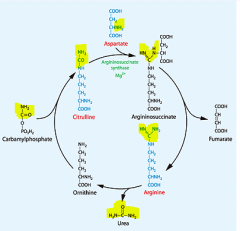
|
|
|
Is nitrogen electron donating or electron pulling from C?
|
donatin
|
|
|
which is more basic then?
arginine vs lysine? |
arginine because it has more electron donation
|
|
|
Are basic AA's positive or negative at body pH?
|
positive because they have so much electrons to give
|
|
|
What are the basic AA's? from most to least bqsic? (3)
|
arginine
lysine histidine |
|
|
What are the acidic AA's?
|
only Aspartate and glutamate
|
|
|
Which AA's are in histones and why?
|
Lysine and arginine because they are most heavily negatively charged so they attract the negative DNA well
|
|
|
What is histidine's charge at body pH?
|
none- neutral
but it is capable of being positively charged |
|
|
What is the alanine cycle?
What is it's counterpart |
an alternate way of getting pyruvate from the muscle to the liver
close to the Cori cycle |
|
|
Why would you need to use the alanine cycle vs the Cori cycle?
What is the burden spared on the muscle cells in each? |
Alanine- breakdown of AA for energy, need to unload the NH3 onto the liver
COri- beakdown of glucose for anaerobic energy, need to unload electrons to be oxidized to the liver (NADH) |
|
|
What is similar in both cycles?
|
they both essentially send some form of pyruvate to the liver for gluconeogenesis
|
|
|
What happens to the inloaded burden products in each cycle?
|
alanine- NH3 disposed of as urea using urea cycle
cori- electrongs sent along ETC |
|
|
What is the intermediate used to transfer NH3 waste products onto pyruvate to make the alanine?
Why is this so perfect? |
a-ketoglutarate takes up NH3 from AA's to become glutamate, which will unload to alanine
aKG is already in the mitochondria (where AA will feel into Krebs) as a TCA intermediate. |
|
|
Why is it so important to attach an amino group to an AA or urea in the body?
|
free floating NH4+ is toxic
|
|
|
Where is the only part of our body where we have free floating ammonium?
|
in the urine excreted out by glutamine to capture and buffer H+
|
|
|
What is the main way nitrogen is exreted in our bodies?
Name 3 alternatives. |
via urea
uric acid, NH4+, creatinine |
|
|
show creatinine
|
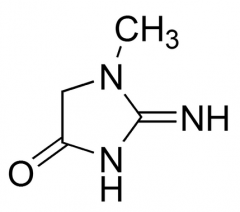
|
|
|
What happens to ingested AA's in excess of what we need for energy and catabolism?
|
it is turned into glycerol and FA's for lipogenesis
|
|
|
Why isn't it turned into glucose?
|
gluconeogenesis is triggered by glucagon and this is probably after a meal, which means that insulin is present
|
|
|
What are the two fates of muscle protein during fasting?
|
sent to liver and made into...
ketone bodies and glucose |
|
|
What are the essential AA's? Mnemonic?
|
PVT TIM HALL
Phenylalanine Valine Tryptophan Threonine Isoleucine Methionine Histidine Arginine Leucine Lysine |
|
|
How do you remember some main ones that are not essential?
|
think glutamate, aspartate, their amine forms
these are obvs synthesized and available all over |
|
|
What 2 AA's do you need more of in growth?
Mnemonic? |
Arginine and Histidine
AH, we are growing! (both basic) |
|
|
From what molecule are the nonessentials made from?
|
glucose
|
|
|
What are the 2 conditionally essential AA? Why?
|
Cysteine because it needs a sulfur group donation from methionine
Tyrosine- made from hydroxylation of phenylalanine |
|
|
From what is uric acid made from?
|
purines
|
|
|
What is the cofactor for transamination reactions?
|
pyridoxal phosphate (PLP)
|
|
|
We get rid of AA aminos via urea. How do we get rid of ammonium ions?
|
place them on purines
|
|
|
What AA often participates in transmination reactions?
|
glutamate/a-ketoglutarate
|
|
|
What are two general ways in which you can hyperammonemia?
|
1. liver disease (no hepatoctes for urea cycle)
2. urea cycle is not working (missing cofactor or enzymes) |
|
|
What gets depleted in ammonium toxicity?
|
a-ketoglutarate which accepts all of it
|
|
|
what happens if a-KG is depleted?
|
no TCA = no energy
|
|
|
What are some sx of ammonium roxicity and explain why they happen?
|
the most energy demanding cells will start to fail first- neurons and eyes
tremor (asterhexis), slurred speech, somnolence (loss of neocortex), vomiting, blurred vision, cerebral edema |
|
|
What is a dietary treatment for hyperammonemia?
|
low protein/nitrogen diet
|
|
|
So what kind of diet is really bad for an alchoholic with cirrhosis?
|
high protein/meat diet
|
|
|
What form of nitrogen can pass and cannot pass cell membranes?
|
NH3 can pass
NH4+ cannot pass |
|
|
What is the only way (generally) that nitrogen can leave the body if the urea cycle is not working?
|
via the gut
|
|
|
What drug utilizes this ion trapping to help with high ammonium?
|
Lactulose- acidifies the GI to trap NH4+ out there
|
|
|
What is another way that nitrogen can be trapped for excretion? (2 drugs)
|
benzoate and phenylbutyrate to bind AA in the GI to prevent absorption
|
|
|
What does N-acetylglutamate look like and how do we get it?
|
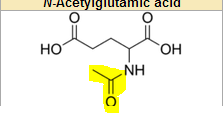
it is synthesized in our body from Acetyl CoA and glutamate
|
|
|
What reaction ceases if we are short on N-acetylglutamate?
Why is it N-acetyl? |
it is connect by an Nitrogen
the first incorporation of NH2 from an AA with CO2 to carbamoyl phosphate |
|
|
How do we get N-acetylglutamate deficiency? How do you know this?
|
we inherit it because we synthesize this
|
|
|
What other ingerited disorder is it similar to? WHy?
|
caramoyl phosphate synthase deficiency- same reaction
|
|
|
How can we tell the two apart?
|
if we see hyperammoniunemia, no urea, and normal urea cycle enzymes, then it is the cofactor
|
|
|
What urea cycle intermediate will be raised in any kind of hyperammonemia?
|
ornithine!
|
|
|
Which part of the urea cycle happens in the mitochondria?
|
the caramoyl phosphate ones (synthesis and incorporation into ornithine transcarbamylase)
|
|
|
What builds up if you have ornithine caramoylase deficiency?
|
the carbamoyl phosphates!
|
|
|
What happens to the caramoylphosphates if they can't become urea?
|
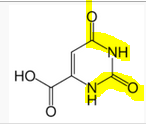
they are fed into the pyrimidine synthesis pathway and excreted as orotic acid
|
|
|
Mnemonic for orotic acid?
|
it is good enough to be ornithine replacement!
|
|
|
What are 2 findings and 1 general sx of ornithine transcarbamylase decifiency?
|
high orotic acid in blood
low BUN sx of hyperammonemia |
|
|
Why would you have hyperammonemia if you are diverting the ammonia to orotic acid?
|
I guess this pathway is not as efficient as the urea cycle
|
|
|
What demographic (age and sex) should I think of with the ornithine transcarbamylase deficiency and why?
|
baby boys
shows early on and is X-linked recessive |
|
|
What are the other urea enzyme deficiencies genetically?
|
autosomal recessive
|
|
|
So which urea genetic deficiency is most common?
|
the X-linked ornithine transcarbamylase deficiency
|
|
|
Now we talk about AA derivatives!
What are the two main functions of BH4? What does this stand for? |
tetrahydrobiopterin
it will hydroxylate aromatic amino acids and make NO |
|
|
What AA is NO made from and why?
|
arginine because it has the most N
|
|
|
What is vitamin B6 also known as? (most famous of the variations)
|
pyridoxal phosphate (PLP)
|
|
|
Show the reaction to make GABA from an AA
What cofactor did this need? |

B6
|
|
|
What does DOPA stand for?
|
di-hydroxy phenylalanine
|
|
|
What other cofactor can add OH, but doesn't do so on an aromatic ring?
|
vitamin C
|
|
|
How do you remember that vitamin C does this?
|
it does this to collagen AA residues to modify them
|
|
|
What is SAM-e? What does it stand for? What is it made from?
What does it do? |
S-adenosylmethionine made from ATP and ATP
it is a cofactor for adding a methyl group to a compound |
|
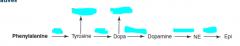
Name the cofactors for each step and their general function.
Also name the two derivatives of tyrosine and DOPA |
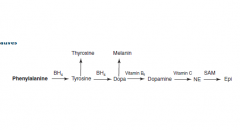
BH4- hydroxylating aromatic rings (x2)
B6/PLP- decarboxylation and trans/deamination Vitamin C- hydroxylation of AA's SAM-e- methylation |
|
|
How do you know melanin is from DOPA synthesis?
What does thyroxine look like? |
In parkinson's, you ose DOPA and the blackness in the SN
Thyroxine looks like 2 tyrosine fused together |
|
|
What is melatonin directly derived from? Steo before that?
|
tryptophan --> serotonin --> melatonin
|
|
|
WHat about eating habits before sleep makes so much more sense now?
|
high carb tryptophan increases not only serotonin, but melatonin
|
|
|
Using knowledge of sleep hormone function, how can you explain my experience with melatonin?
|
the sleepiness passes quickly because there is only a surge of serotonin present to get you to sleep in the first place
|
|
|
What cofactor does COMT have?
|
SAM-e
|
|
|
What does tryptophan look like?
|
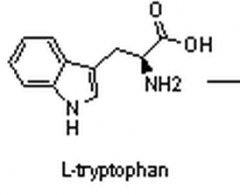
|
|
|
What is the precursor to serotonin?
|
5-HT
|
|
|
What does 5-HT stand for?
|
5-hydroxytryptophan
|
|
|
Imagine the structural changes from tryptophan to serotonin.
|
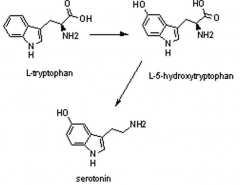
|
|
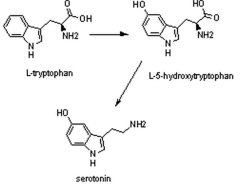
What cofactor is used in each step?
|
Trp --> 5-HT = BH4
5-HT --> Serotonin = B6 |
|
|
How is serotonin modified to become melatonin?
cofactor? |
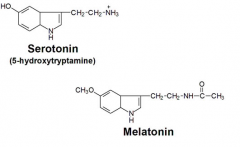
Acetylated
COMT attaches a methyl to it's aromatic OH SAM-e |
|
|
What does this explain about COMT inhibitor side effects?
|
why you may have sleep disturbances taking them
|
|
|
What AA is used to make porphyrin?
How many subunits and of what is in it? |
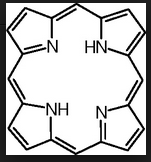
glycine
4 pyrrole rings |
|
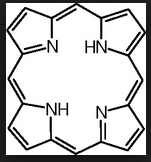
What cofactor must be necessary to turn glycine into porphyrin?
|
B6 because are the carboxylate groups are gone
|
|
|
How does heme and cytochrome look different than porphyrins?
|
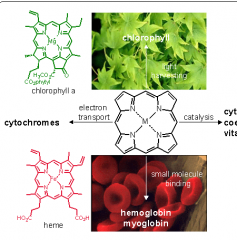
appendages and metals added
|
|
|
Two things glutamate can be made into
|
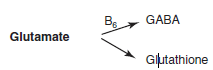
|
|
|
3 things arginine can be made into
|
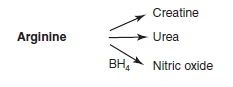
|
|
|
composition of glutathione?
|
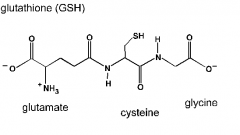
glutamate, cysteine, and glycine
|
|
|
How is BH4 made?
What does it do again? |
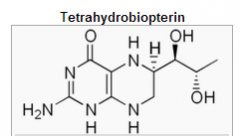
hydroxylation of aromatic AA's
production of NO made from GTP (looks like a purine!) |
|
|
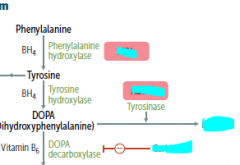
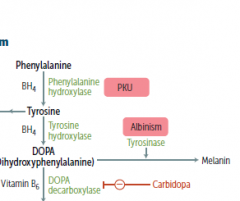
What happens if Phenylalanine cannot be synthesized to tyrosine?
|
you get phenylketouria (PKU)
|
|
|
Two ways in which you can be born with PKU?
|
no phenylalanin hydroxylase enzyme
no ability to synthesize BH4 from GTP |
|
|
would a person with PKU suffer from depression and other catacholamine deficient sx? Why?
|
no, because they will still get tyrosine in their diet to make this
only worry is Phe building up |
|
|
What happens to Phe when it builds up? Reaction and products?
mnemonic? |
it is deaminated to a phenylKETOne that is excreted in the urine because we can't use any more of it.
puts the K in phenylketouria! |
|
|
What are the 3 phenylketones?
|
phenylacetate, phenyllactate, and phenyl pyruvte
|
|
|
What does phenylacetate/phenylpyruvate look like?
What main reaction has happened? cofactor for both? |
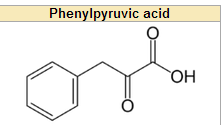
literally just a acetate/pyruvate attached to a phenyl ring
deamination for phenylpyruvate = B6 decarboxylation of PP for Pacetate = B6 |
|
|
What kind of real and special foods must PKU patients have in their diet?
|
No real food proteins (prob have Phe), but can have starch and veggies (no meat, dairy, or beans)
Special shake of AA's, vitamins, and minerals. |
|
|
What AA's do you aim to increase vs decrease in PKU?
|
increase tyrosine
decrease phenylalanine |
|
|
How common is PKU and how is it commonly diagnosed?
|
1 in 10,000
every newborn is screened for it |
|
|
What artifical sweetener must PKU pts avaoid? why?
|
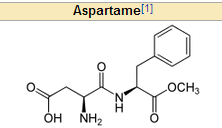
aspartame because it has Phe
|
|
|
What 2 AA's is aspartame made of? Mnemonic?
|
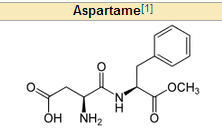
PKU can't have it and ASPARTame!
Phenylalanine and aspartame! |
|
|
Are ketones or amine more volatile? Why?
|
ketones - they don't have hydrogen bonds to hold them down
|
|
|
What smell do PKU pts have and why?
|
musty odor because the pheynketone metabolities become volatile from the sweat and urine
|
|
|
What is a sx of untreated PKU in baby's skin? Why?
|
fair skin because they can't make melanin as well
|
|
|
What are some imediate effects of ingesting Phe not counting the smell? Why?
|
brain damage: seizures, mental retardation
PK are readily taken up into the brain |
|
|
What is maternal PKU?
|
when a mother with PKU does not manage their diet properly and the infant is esposed to a lot of PK's
|
|
|
Why is it especially bad for the fetus?
Is the woman at any more risk than normal? |
because the fetus is most definitely a carrier
|
|
|
What are 3 sx of maternal PKU in the infant?
|
microcephaly, growth retardation, congenital heart defects
|
|
|
|
|
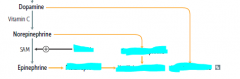
degradation products
|
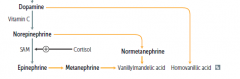
|
|
|
Why does it make sense that cortisol enhances NE --> Epi?
|
both are secreted in the adrenal medulla in times of stress.
cortisol travels inward and increases enzymes for NE methylation |
|
|
dissect the word alkaptonuria
|
alka- alkali
uria- in urine something basic in the urine |
|
|
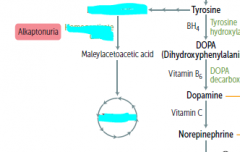
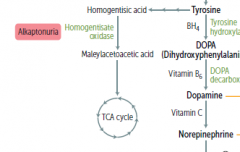
What process is wrong in alkaptonuria?
|
the degredation of Phe and Tyr is impaired..
|
|
|
What builds up in alkaptonuria?
|
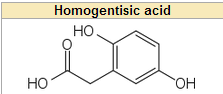
an intermediate metabolite called homogentisic acid
|
|
|
What is the main sign of alkaptonuria? alternate name?
|

black urine disease
|
|
|
|
|
|
mnemonic for component built up in alkaptonuria!
|
a black homosexual gentleman is fantastic!
black urine homo-gent-tistic |
|
|
What do pt's with alkaptonuria look like on the otside? Why?
|
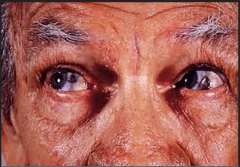
pigmented sclera and skin because of buildup of homogenistic acid
|
|
|
What kind of sx will these pts have?
Is it generally considered fatal? |
no, it's relatively benign
but the stuff will attach to catilage and cause achey joints |
|
|
What will their CT look like?
|
brown/black
|