Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
28 Cards in this Set
- Front
- Back
|
Describe the differences in aetiology of immunodeficiencies and haematopoeitic malignancies
|
- Primary immunodeficiency and haematopoietic malignancies are related in both being defects of leukocyte development during haematopoiesis
- In both there is a failure of cells to develop to maturity - Immunodeficiency = the defect prevents proliferation and/or function - In malignancy the defect results in excessive proliferation of (usually) non-differentiated cells which are clonal in origin - Because of this close relationship, malignancies often develop in individuals with immunodeficiency (and autoimmunity, which may also be a sort of developmental deficiency of the haematopoietic system) and vice-versa |
|
|
What are primary immunodeficiencies (PIDs)?
|
- Intrinsic defects of the haematological system, most likely due to genetic factors
- These may be inherited - Or sporadic (i.e. arising either in parental gametes or at some stage of development) - mutation is more common |
|
|
What are secondary immunodeficiencies (SIDs)?
|
- Due to extrinsic factors e.g.
• Infection e.g.. AIDS • certain drugs or chemical agents e.g. immunosuppresants given to transplant patients, and anti-cancer drugs • Other underlying diseases |
|
|
Describe the characteristics of primary immunodeficiencies
|
- Failure of one or more branches of the immune system
- Higher rates of autoimmune diseases - Enhances susceptibility to infection - Increased rates of some kinds of cancer especially haemtological (solid tumours may themselves be immunosuppressive in advanced stages) - Primary immunodeficiencies is uncommon (120 primary diseases described) and patients need life-long management |
|
|
Describe the treatment for primary immunodeficiencies
|
- Symptomatic e.g. steroids for autoimmunity (e.g.. thrombocytopnaea)
- Prophalaxis against infection = antibiotics (for life), pooled human Ig given IV (IVIG) (inject Abs into patient) - Bone marrow/stem cell transplant = replace immune system by repopulating bone (curative but may lead to Host-vs-graft disease) - Gene therapy = take bone marrow stem cells, replace faulty genes and reintroduce them (may lead to leukaemia) |
|
|
Describe the causes of secondary immunodeficiencies
|
- Very common = AIDS about 60000 (1:1000) living with HIV in UK who would become immunodeficient without treatment
- Immunosuppression to treat autoimmunity and for transplant - Other drugs e.g. cancer chemotherapy which causes myelosuppression - Splenctomy e.g. due to rupture because of trauma → spleen clears bacteria from the blood so very important - Some cancers |
|
|
Describe 5 features of immunodeficiencies that may lead to diagnosis
|
1. Failure to thrive in babies = failure to maintaining a normal rate of growth such that the baby's rate falls significantly behind that of a normal childs
2. Enhanced susceptibility to infection 3. Jaundice, diarrhoea, rashes 4. Associate features of specific syndromes (e.g. autoimmunity cardiac defects, abnormal appearance e.g. in DiGeorge syndrome) 5. Predisposition to some cancers |
|
|
How is a diagnosis of immunodeficiency confirmed?
|
1. Through lab demonstrated reductions in the functionality of the immune systen e.g. lowered Ig
2. Demonstration of genetic defects |
|
|
What is the main defining clinical feature of immunodeficiency?
|
- Infections of all sorts
- Important that the infections are: 1. Serious = infections which may normally be trivial but which may lead to life-threatening in IDs e.g. chicken pox in leukaemia patients 2. Persistent e.g. Candida (thrush) infections, enteroviral (gut) infections 3. Unusual = opportunistic infections that do not usually present symptoms e.g. Pneumocystis jiroveci in AIDs 4. Recurrent e.g. diarrhoea, lung infections - Remember SPUR |
|
|
Describe how the type of infections may show which component of the immune system are faulty
|
- Different parts of the immune system are designed to fight different sorts of infection, and are therefore indicative of non-functioning if the prevalence of these infections increases (but not diagnostic)
- Antibody = mainly bacterial (and helps against viruses) - T-cells = fights against bacteria, viruses and fungi - Complement = mainly bacteria but helps against viruses - Neutrophils = bacteria and fungi |
|
|
Describe the features and diagnosis of immunoglobulin deficiencies
|
- These present with upper and lower respiratory infections
- Often not considered until many infections and organ damage (e.g. bronchiectasis, deafness) - Simple to test for = measure immunoglobulin levels - However, must take account of patients age = neonates have only IgG from mother and blood concentrations naturally decline over first 1-2 months and then rise to adult levels over years |
|
|
Describe 3 examples of primary immunoglobulin deficiencies
|
1. Bruton's X-linked agammaglobinaemia (XLA)
• Mutation of locus for a tyrosine kinase needed for pre-B cell proliferation on the X chromosome • Affects infant boys to adults with variable severity • Produce no B cells (CD19 sIg), so no Ig • Other immune functions ok • IVIG treatment leads to near-normal life-span • Very rare • Arthritis and IBD occurs 2. Common variable immunodeficiency (CVID) (AKA non-familial hypogammaglobinaemia) • Very heterogenous (variable) • About 10% familial, the rest with unknown aetiology • Reduced IgG, IgA and often IgM (may have all 3) • Susceptible to bacteria but not viruses • B cells are normal but plasma cell differentiation defective and class switching does not occur • May be a Th defect rather than a specific B-cell defect • Arthritis and other AI diseases common • Lymphoma the commonest cause of death • For diagnosis first search for specific IgG antibody levels = will all be undetected, even if recent boosters • Diagnosis usually by elimination of other possibilities → AIDS should always be considered → SCID = consider age and appearance of T-cells → Malignancy e.g. lymphoma, myeloma needs to be eliminated • Treatment generally maintenance = IVIG and prophylactic antibiotics • CVID is usually a relatively mild and treatable condition so a stem cell transplant or gene therapy is not done (too dangerous) • Must watch out for cancer 3. Subclass deficiency • IgA deficiency mostly asymptomatic (may affect mucosal immunity) • IgG2 deficiency predisposes to lung infections especially with encapsulated bacteria • Again, AI diseases occur |
|
|
Give 2 examples of secondary imunoglobulin deficiencies
|
1. Nephrotic syndrome = renal damage leading to proteinuria and so hypogammaglobinaemia
2. Chronic B lymphocytic leukaemia (CLL) = over-production of immature B-cells leading to hypogammaglobinaemia (over some years) |
|
|
Describe the features of T cell deficiencies
|
- These inevitably are very serious because T cells are essential for all branches of the adaptive immune system and without them immunodeficiency is profound = no adaptive immune system
|
|
|
Give 2 examples of primary T cell deficiencies
|
1. Severe combined immunodeficiency - SCID
• Untreated results in death, usually within weeks of birth from overwhelming infection = very severe • All branches of the immune system are affects = combined • Problem in SCID is the development of lymphocytes = always T cells, often B cells and/or NK cells also (depends on how far along the haematopoietic pathway the defect is) • This leads to failure of all branches of the adaptive immunity as Th cells are essential • About a dozen genes are identified involved with SCID = X-linked commonest (50%), of which 30% is inherited; the rest are autosomal recessive • The genes affected are involved with: → Aspects of cell signalling essential for T cell development and function e.g. IL7R (crucial developmental cytokine) → T cell receptor arrangement, essential for T cell development e.g. RAG1 (crucial for generation of the receptor) → Removal of toxic metabolites e.g. adenosine deaminase (ADA) • SCID is suggested in an infant by repeated severe infections, particularly of bowel and upper respiratory tract by common patholgens and by opportunistic infections (e.g. Aspergillus spp.). These cause failure to thrive • SCID is confirmed by absence (or very reduced numbers) of a least T cells - but note that occasionally T cell numbers are normal but the cells are non-functional • Suspect SCID in any neonate or infact with FTT, infections and recurrent hospital admissions and call an immunologist • Treatments - Maintenance = barrier nursing and prophylactic antibiotics, IVIG, no live vaccines and no whole blood infusions - Curative = allogeneic stem cell CD34 cell transplant or gene therapy (in vitro transformation of patient's CD34 cells then re-infusion) → only successful example of gene therapy 2. DiGeorge syndrome • Congenital but not familial • Due to a deletion in chromosome 22, at a hot spot in very early stage of development → a developmental problem leading to thymic hypoplasia so few T cells (also cardiac and facial problems) • A similar syndrome may be associated with foetal alcohol syndrome |
|
|
Give 2 examples of secondary T cell deficiencies
|
1. HIV infection
2. Iatrogenic (e.g. anti-T cell Ab therapy) |
|
|
Describe the features of complement deficiencies
|
- Affect either the classical pathway of the alternate pathway
• Classical pathway activated by Ag-Ab complexes (adaptive) → bacterial infections, immune complex diseases • Alternate pathway activated either by bacterial mannans via mannan binding lectin (MBL) or by other cell surface components binding complement → meningococcal and gonococcal infections - Complement deficiencies due to a lack of one of the many components of the complemen system - Nearly all are due to genetic defects - Consequences: • Infections, due to failure of Ab-mediated complement lysis of bacteria, especially Strep. pneumoniae, Neisseria meningitides • Immune complex diseases because of failure to clear Ag:Ab complexes e.g. SLE, glomerulonephritis - Diagnostic tests: • Complement function = classic haemolytic 50 (CH50) measures activity of the classical pathway, AP50 measure activity of alternate pathway → titrate serum: determine concentration to lyse 50% of red blood cells (coated with Ab in CH50) • Absence of specific components e.g. ELISA for MBL |
|
|
Give 3 examples of primary neutrophil defects
|
1. Chronic granulomatous disease (CGD)
- Defect of neutrophil killing of phagocytosed organisms, especially catalase positive organisms - Neutrophil numbers normal - X-linked (more common in males) - Extremely rare, but almost as severe as SCID - Due to an X-linked deficiency in NADPH oxidase = the enzyme that powers the 'oxidative burst' in phagocytic cells which kill bacteria, and so defect leads to failure to kill phagocytosed pathogens - Patients present with: • Severe infections and formation of abscesses = commonly Staph. aureus • GI inflammation • Delayed wound healing with granulomas • Survival into 20s rare until recently - Neutrophil numbers normal so test for neutrophil function of oxidative burst = NBT test (cells go blue when oxidative burst stimulated) - Treatment • Prophylactic antibiotics • Leukocyte infusions • Bone marrow transplant • Interferon gamma = cytokine activating oxidative burst (v. expensive) 2. Congenital neutropenia 3. Leukocyte adhesion deficiency |
|
|
Give 3 examples of secondary neutrophil defects
|
1. Neutropenia due to cancer chemotherapy
- Chemotherapy is myelosuppressive = bacterial infections in neutropenic cancer patients was a major cause of death until recently - A fever in neutropenic patientis is a major emergency 2. Myeloid leukaemia 3. Late feature of liver failure |
|
|
What are haematopoietic malignancies?
|
These are a large and very diverse group of diseases - but only 6 occur frequently
- underlying defect is uncontrolled proliferation of a lymphoid or myeloid precursor cell (i.e. not mature so not functional) OR a more mature cell including B lymphocytes or plasma cells; - lead to defects in the immune system (but this is usually not the patient’s main problem). |
|
|
What are the 3 main types of haematopoietic malignancies and how are they subdivided?
|
1. Leukaemia
- cells proliferate in the marrow & immature cells spill over into circulation (hence leuk | aemia); 2. Lymphoma - mature lymphocytes proliferate in lymph nodes which swell (hence lymph | oma), not present in circulation; 3. Myeloma - plasma cells proliferate in bone marrow (myel | oma), not present in circulation. - Each type is then named for the specific cell type involved e.g. B lymphoma affacting mature B cells e.g. acute myeloid leukaemia affecting pre-granulocyte/monocyte cells |
|
|
Describe the development of haematopoietic malignancies
|
- May develop rapidly = weeks e.g. acute leukaemias and high grade lymphoma
- May develop slowly (years) e.g. chronic leukaemias e.g. CLL and low grade lymphomas = but both eventually transform into more severe versions similar to the acute/high grade disease |
|
|
Describe the diagnosis of haematopoietic malignancies
|
- Constitutional symtoms:
• high grade lymphomas/acute leukaemias: weight loss, malaise, fevers, night sweats, swollen lymph nodes: and myelosuppression, leading to neutropenia ( → infections) thrombocytopenia ( → bleeding) and anaemia (→ respiratory distress). • low grade leukaemias/chronic leukaemias: often asymptomatic, discovered during other investigations. - Diagnosis: • Presence of excess/abnormal cells in blood/marrow/lymph nodes = blood smears/biopsy • Immunophenotyping a major diagnostic tool i.e. identification of surface markers (CD …) and identification of specific somatic genetic defects e.g. Philadelphia chromosome (t9:22). Important to identify genetic defect to help predict how aggressively the cancer will progress and how to treat it |
|
|
Describe immunodeficiencies in haematopoietic malignancies
|
- Secondary immunodeficiency is prominent in AML, CLL, myeloma
• patients may present with repeated infections much as in other immunodeficiencies. - This occurs because: • normal haematopoiesis is disrupted due to malignant stem cells displacing normal haematopoietic stem cells from their bone marrow niches; • the structure of 2ndry lymphoid tissues is disrupted; • the malignant cells are non-functional because not fully differentiated • malignant lymphocytes are monoclonal (like most cancers) so produce no useful antibodies/T cell function. |
|
|
Describe the features of myeloma
|
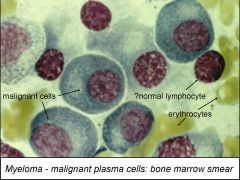
- Often referred to as multiple myeloma, aka plasmacytoma, and there are several related “pre-malignant” conditions.
- Malignancy of plasma cells, developing in marrow but may spread to soft tissues. - Myeloma is due to the uncontrolled clonal expansion of a plasma cell. - This means that a single activated B cell has differentiated to a plasma cell which normally should die (apoptosis) but this one does not: instead it proliferates. - Normal mature B lymphocytes makea unique Ig (sIg, not yet secreting, not a plasma cell). - In Normal B cell activation (reactive): each separate B cell is stimulated to proliferate & become plasma cell, so multiple clones, hence polyclonal immunoglobulin in blood. - Myeloma: one single B cell proliferates to form a plasma cell clone: this produces a unique monoclonal antibody in blood. - The monoclonal immunoglobulin product of myeloma cells is described as paraprotein • may be intact immunoglobulin (e.g. IgG2) or fragments (e.g. free light chain) • Detected in serum by electrophoresis • normal polyclonal Igs form a smear because different Igs have different mobilities; • unique paraprotein forms a tight band. • Free light chains can pass into urine = called Bence Jones protein. - Cytokine production by myeloma cells activates osteoclasts resulting in bone resorption, hypercalcaemia & so fractures. - Deposition of L chains as amyloid in kidney can lead to kidney failure. |
|
|
Describe the diagnosis of myeloma
|
Presence of 2 of 3 features:
- Serum and / or urine paraprotein - Osteolytic lesions on x-ray - >25% plasma cells in marrow (or clonal population) (this is very difficult to do accurately from a bone marrow smear). |
|
|
Describe the treatment of myeloma
|
- Chemotherapy with cytotoxic drugs
• high dose chemotherapy with bone marrow/stem cell transplantation - bisphosphonates to slow bone resorption - dialysis for kidney failure (but not suitable for kidney transplant) - Sadly myeloma cannot be cured so the purpose of the treatment is to relieve symptoms and prolong life = especially bone pain and fracturing |
|
|
What are the main differences between lymphoma/leukaemia and myeloma?
|
Lymphoma / leukaemia
- related to developmental stage of leucocyte Myeloma - clonal plasma cells, secreting monoclonal immunoglobulin (paraprotein) - diagnosed by marrow and serum / urine |