Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
48 Cards in this Set
- Front
- Back
|
Which of the following statements are correct regarding refractory anemia with ringed sideroblasts (RARS)?
a. It is commonly caused by parvovirus infection. b. Bone marrow is hypocellular and shows depletion of the erythroid series. c. Abundant hemosiderin-laden macrophages are present in the bone marrow. d. Evolves to acute leukemia in 50% of cases. e. It characteristically occurs in children. |
c. Refractory anemia with ringed sideroblasts
(RARS) characteristically affects older individuals and shows evidence of iron overload in the bone marrow, spleen, and liver. It is an idiopathic disease that can only be diagnosed after excluding all other possible etiologies of erythroid abnormalities. It evolves to acute leukemia in 1% to 2% of cases. |
|
|
BCR/ABL gene fusion at the major breakpoint cluster region leads to all of the following events EXCEPT:
a. Clonal hematopoietic cell proliferation b. Downregulation of tyrosine kinase activity c. Enhanced transcription of MYC and BCL-2 genes d. The formation of a p210 protein e. Chronic leukemia |
b.In chronic myelogenous leukemia
, the breakpoint in the BCR gene is in the major breakpoint cluster region (BCR exons 12-16) and a p210 protein is formed. The fusion gene leads to increased tyrosine kinase receptor activity. Inhibition of tyrosine kinase by Gleevec is a mode of treatment of chronic myelogenous leukemia. |
|
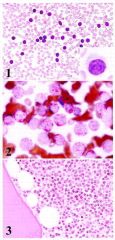
Positive stains: CD19, CD20, CD22, DBA.44, CD79a, PAX5, TRAP (tartrate resistant acid phosphatase); also CD11c, CD25, CD103, HC2, FMC7; reticulin shows thickened fibers that may circle individual cells and infiltrate normal appearing areas
|
Hairy cell leukemia
|
|
|
GASTRIC MALTOMA
|
•antibiotic therapy often (>70%)
induces regression (unless t(11;18)+) |
|
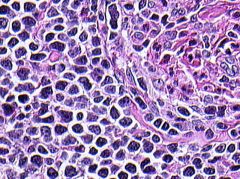
Positive stains: CD5, CD19 (strong), CD20 (strong), cyclin D1/bcl1 (variable nuclear staining since cells are at different stages of cell cycle); also CD22, CD43, CD79a, FMC7, surface IgM or IgD, kappa or lambda, bcl2
In-situ hybridization for bcl-1 is more sensitive/specific than immunostains |
Mantle cell lymphoma
|
|
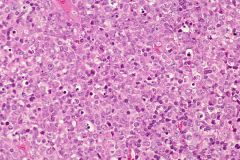
Young women with cervical lymphadenopathy (tender or painless), fever, often leukopenia
|
Kikuchi’s disease in lymph nodes
|
|
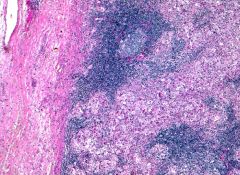
Massive painless bilateral lymph node enlargement in neck, of unknown etiology
Associated with fever, leukocytosis, anemia, elevated sedimentation rate, polyclonal hypergammaglobulinemia Usually age 20 years or less, but can affect any age |
Sinus histiocytosis with massive lymphadenopathy
Positive stains: S100, CD68; also CD30, CD14, oil red O Negative stains: CD1a, EBV, CD21 |
|
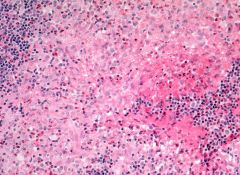
|
Langerhans cell histiocytosis in lymph nodes
Positive stains: CD1a, S100; also vimentin, langerin, fascin (AJCP 2002;118:335), CD68, CD74, HLA-DR, CD45 (at frozen section); variable CD2 and CD3 Negative stains: CD15, CD45RA, CD45RB, EMA, CD21, CD35 |
|
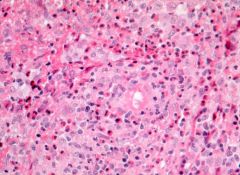
|
Langerhans cell histiocytosis in lymph nodes
Hand-Schuller-Christian’s disease: term used previously for more indolent disease of children and young adults Hashitmoto-Pritzker’s disease: congenital, self limited form Letterer-Siwe’s disease: systemic disease in infants |
|
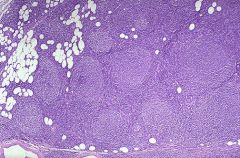
Most common form of non-Hodgkin lymphoma in US (45% of all cases), rare in Asians, African-Americans
Median age 55 years; rare under age 20 Generalized painless lymphadenopathy and marrow involvement (85% have paratrabecular lymphoid aggregates), 10% with peripheral lymphocytosis; occasional involvement of splenic white pulp and hepatic portal triads |
Follicular lymphoma
|
|
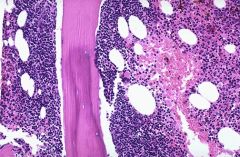
|
Grading: 1, 2 or 3 based on number of intrafollicular centroblasts in ten 40x fields to determine average (WHO)
Follicular lymphoma |
|
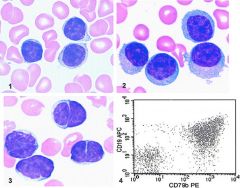
Often presents with leukemia although patients may be asymptomatic
Median survival 4-6 years; indolent but incurableUsually older patients (median age 60), 2/3 male, with disease in bone marrow, lymph nodes, spleen, liver |
CLL/SLL
Poor prognostic factors: 17p deletions, 11q22-23 deletion, non-mutated immunoglobulin genes, abherrant expression of CD2, CD7, CD10, CD13, CD33 or CD34 Positive stains: CD5, CD19, CD20 (dim), CD23, surface Ig light chain, surface IgM (dim); also CD43, CD79a, CD79b (dim, in 20%), bcl2; variable CD11c, FMC7 (42%) Negative stains: CD10, cyclin D1 |
|
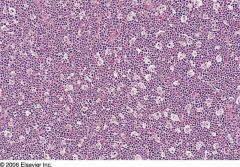
|
Burkitt’s lymphoma
30% of childhood lymphomas, may present as B cell acute lymphoblastic leukemia FAB L3 (also called Burkitt’s leukemia) Either endemic, sporadic or immunodeficiency-associated In adults, often associated with immunodeficiency, including HIV; involves distal ileum, cecum, mesentery In tropical Africa, involves jaw or abdomen (endemic), 95% are EBV positive; malaria may facilitate EBV infection Curable with aggressive therapy in 60% Due to c-myc translocation that causes increased constitutive levels of c-myc |
|
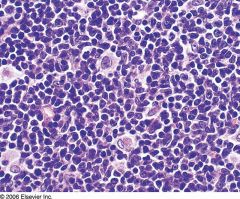
Formerly known as centrocytic lymphoma
3-10% of non-Hodgkin lymphomas in Western countries Usually 60+ years with stage III/IV disease; 80% men Lymphadenopathy, bone marrow involvement in 2/3 (lymphoid aggregates, often paratrabecular), splenomegaly in 50% with white pulp involvement, periportal hepatic infiltration, lymphomatoid polyposis of small bowel and other extranodal involvement; CNS involvement in 10%; B symptoms (fever, night sweaters, weight loss) in 1/3 |
Mantle cell lymphoma
|
|
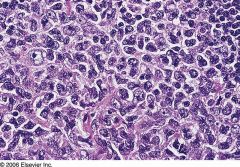
All ages, 30%-40% are extranodal (skin, bone, gastrointestinal, genitourinary, CNS); 50% are stage III/IV at diagnosis
Often single, rapidly growing mass; may be in liver, spleen; bone marrow involvement typically late Fatal if untreated; remission in 70% after chemotherapy, 50% cure rate |
Diffuse large B cell lymphoma
|
|
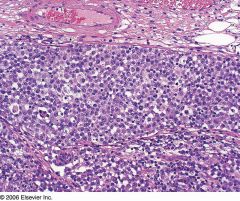
Also called Ki-1 (CD30+) lymphoma
3% of adult and 10-30% of childhood non-Hodgkin lymphomas Usually T-cell origin and T-cell lineage, which can usually be proven by molecular studies, even if no T-cell antigen expression Rarely null cell types; younger than T cell types (28 vs. 42 years, |
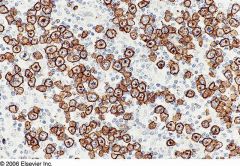
Anaplastic Large Cell Lymphoma
Strong membranous and Golgi-type immunoreactivity for CD30 in anaplastic large cell lymphoma. |
|
|
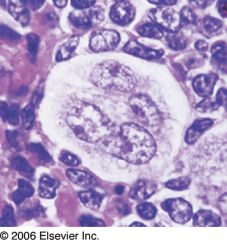
Nodular Sclerosis Hodgkin's lymphoma
|
|
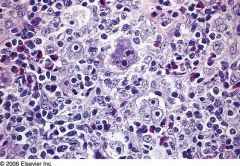
|
Mixed Cellularity Hodgkin's Lymphoma
Large number of: eosinophils plasma cells atypical mononuclear cells Classic Reed–Sternberg cells |
|
|
Progressive transformation of germinal centers is associated with which of the following conditions?
a. Measles b. Toxoplasmosis c. Syphilitic lymphadenitis d. Nodular lymphocyte-predominant Hodgkin disease e. Castleman disease |
d. Progressive transformation of germinal centers
describes an alteration of lymphoid architecture in which lymphoid follicles become enlarged and infiltrated by small lymphocytes. It occurs in association with nodular lymphocyte-predominant Hodgkin disease and other reactive lymphadenopathies. |
|
|
“Smoldering myeloma” is designated as such because of:
a. Absence of lytic bone lesions b. The presence of anemia with low erythropoietin level c. Plasma cells in the bone marrow do not have atypical or pleomorphic forms d. Characteristic M-protein spike in the serum e. Absence of Bence-Jones proteinuria |
b. Smoldering myeloma
patients are asymptomatic, without bone lesions and are usually not treated until they develop symptoms. They have M-protein peak in the serum and plasma cells in the bone marrow and may have Bence-Jones proteins in the urine. They do not have anemia, hypercalcemia, or renal insufficiency. |
|
|
The blasts in this bone marrow of a 17-year-old patient with trisomy 21 will most likely stain with:
a. Tdt b. Glycophorin A c. CD61 d. Myeloperoxidase e. Sudan Black B |
c. Down syndrome patients develop
acute myeloid leukemia of the megakaryocytic type. The blasts are usually MPO, SBB, and Tdt negative and are positive for CD61 (platelet glycoprotein IIIa) and CD41 (platelet glycoprotein IIb/IIIa). |
|
|
Translocation t(9;14)(p13;q32) is identified in cases of:
a. Splenic marginal zone lymphoma b. Extranodal margin zone B-cell lymphoma c. Mucosa-associated lymphoid tissue (MALT lymphoma) d. Lymphoplasmocytic lymphoma |
d. Translocation t(9;14)(p13;q32) and rearrangement of the PAX-5 gene are reported in up to 50% of lymphoplasmocytic lymphoma.
|
|
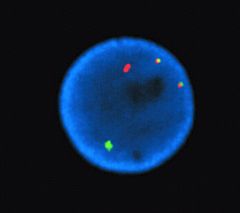
A 19-year-old patient with orbital mass had increased blasts in the bone marrow (15%). A cytogenetic preparation of the bone marrow was studied by in situ hybridization using probes from chromosome 21 and 8 (red and green, respectively).The diagnosis is most likely:
a. Acute myeloid leukemia b. Acute lymphoblastic leukemia/lymphoma c. Myelodysplastic syndrome d. Chronic idiopathic myelofibrosis e. Nonhematologic malignancy |
The image of an interphase nucleus shows fusion transcript of AML1 on chromosome 21 and ETO gene on chromosome 8. Translocation t(8;21)(q22;q22) with AML1/ETO gene fusion is a characteristic finding in
acute myeloid leukemia and patients may be diagnosed with leukemia despite blast numbers of less than 20%. This type of leukemia predominantly occurs in young patients and may present with a myeloid sarcoma tumor. |
|
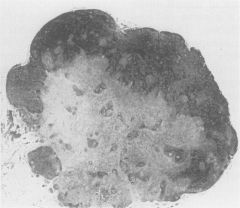
|
Inflammatory pseudotumor in lymph nodes
|
|
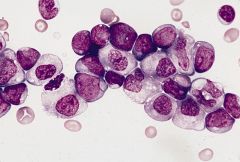
AML1-ETO leukemia
fusion product of ETO gene on #8q22 and AML1 gene on #21q22 5-10% of AML, 33% of cases of AML-M2 (AML with maturation); most common type of childhood AML |
AML with t(8;21)(q22;q22) large blasts, abundant basophilic cytoplasm, frequent large Auer rods and chunky cytoplasmic granules, perinuclear hofs, neutrophil dysplasia; trilineage dysplasia present in therapy related cases
|
|
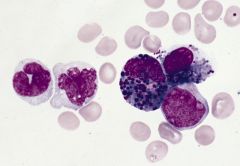
M4Eo; 8% of adult AML, 25% of acute myelomonocytic leukemia (AML M4) cases
Associated with more frequent hepatosplenomegaly, lymphadenopathy and granulocytic sarcoma than AML in general Longer median survival than other AML |
AML with inv(16)(p13;q22) or t(16;16)(p13;q22); granules are positive for chloroacetate esterase and nonspecific esterase; high Ki-67, CBFbeta-SMMHC (nuclear stain with microgranular or fine-speckled pattern)
usually AML M4 (acute myelomonocytic leukemia) features plus marrow eosinophilia with dysplastic eosinophils containing large basophilic staining granules in addition to usual eosinophilic granules; usually >10% dysplastic forms in at least one lineage |
|
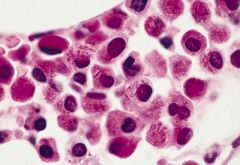
Either hypergranular (this section) or microgranular (section below)
8% of AML cases, 15% of adult AML Formerly called AML M3Median age 35-40 years |
Acute promyelocytic leukemia (APL) with t(15;17)(q22;q12)
|
|
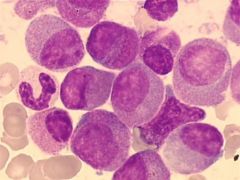
“variant” APL without further description may mean microgranular (morphologic) variant or cytogenetic variant other than t(15;17)
Uncommon, involves retinoic acid receptor alpha on #17 but not PML gene on #15 t(11;17) is most common DIC common |
Acute promyelocytic leukemia with t(V;17)(V;q12); features are intermediate between hypergranular acute promyelocytic leukemia (M3) and acute leukemia with maturation (M2) - most cells have many granules, usually no Auer rods, regular nuclei but increased pseudo Pelger-Huet cells
|
|
|
3-5% of AML
Micro: blasts resemble monoblasts, monocytes or myelomonocytes (AML M4, M5) Positive stains: nonspecific esterase |
AML with 11q23 (MLL) abnormalities
|
|

3-5% of AML
Micro: blasts resemble monoblasts, monocytes or myelomonocytes (AML M4, M5) Positive stains: nonspecific esterase |
AML with 11q23 (MLL) abnormalities
|
|
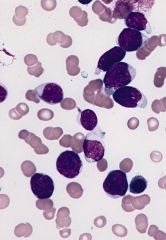
Typically presents with thrombocytopenia, neutropenia and marrow failure
No definitive evidence of myeloid differentiation by morphology and cytochemistry; need immunohistochemistry or EM to characterize as myeloid |
Acute myeloblastic leukemia, minimally differentiated (M0)
Criteria for diagnosis: nongranular blasts; less than 3% of blasts are positive for myeloperoxidase or Sudan Black B by enzyme cytochemistry |
|
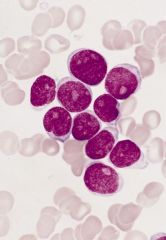
10-20% of AML cases, 44% in one Brazil hospital
|
Acute myeloblastic leukemia without maturation (M1)
Criteria for diagnosis: 90% of nonerythroid cells in marrow are myeloblasts; if no Auer rods, at least 3% of blasts must be positive for myeloperoxidase or Sudan Black B by enzyme cytochemistry |
|
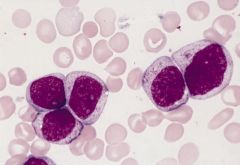
30-45% of AML cases; 5% of childhood leukemias (Orphanet)
Any age, 20% are < 25 years and 40% are 60 years+ Variable prognosis |
Acute myeloblastic leukemia with maturation (M2)
Criteria for diagnosis: 20%+ nonerythroid cells in peripheral blood or bone marrow are myeloblasts; monocytic precursors are < 20%, granulocytes are >10% of cells |
|
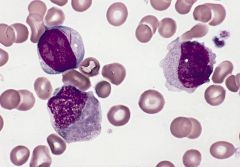
myeloblast (upper left) with two long slender Auer rods, neutrophilic myelocyte (below myeloblast) with smudged nonspecific granules and promonocyte (right) with abundant azurophilic granules and nucleus with delicate folds and creases
|
Acute myelomonocytic leukemia (M4)
Criteria for diagnosis: myeloblasts, monoblasts and promonocytes are 20% or more of nonerythroid cells; myeloblasts and granulocytes are 80% or less of nonerythroid cells; monocyte lineage cells are 20% or more of nonerythroid bone marrow cells If less than 20% of bone marrow cells are monocyte lineage, still M4 if blood monocyte count is 5000/mm3 or more |
|
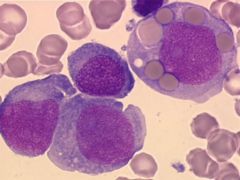
10% of AML cases
High incidence of bleeding disorders (including DIC), organomegaly, lymphadenopathy, gingival hyperplasia, CNS and other tissue involvement (monocytes infiltrate) |
Acute monoblastic and acute monocytic leukemia (M5)
Criteria for diagnosis: 80% or more nonerythroid bone marrow cells are monocyte lineage |
|
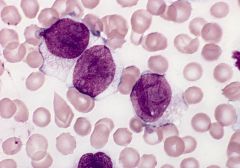
blood smear (Wright-Giemsa): two monoblasts (left) have abundant cytoplasm with numerous azurophilic granules, promonocyte (right) has abundant cytoplasm with fine azurophilic granules, nuclei has delicate folds
|
Acute monoblastic leukemia (M5a)
Criteria for diagnosis: 80% or more monocyte lineage cells are monoblasts |
|
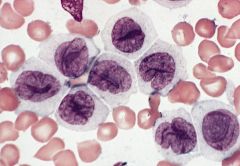
blood smears (Wright-Giemsa): promonocytes have abundant cytoplasm with azurophilic granules that are myeloperoxidase negative, nuclei have delicate folds, nucleoli are inconspicuous
3-6% of AML; affects all ages Mature monocytes or promonocytes predominate in peripheral blood (<80% of monocyte lineage cells are monoblasts, usually <20%) |
Acute monocytic leukemia (M5b)
Micro: leukemic cells are often promonocytes with less basophilic cytoplasm and more azurophilic granules than monoblasts; have folded or cerebriform nuclei with fine chromatin; erythrophagocytosis is common |
|
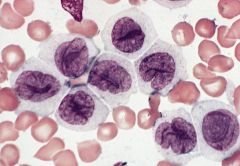
blood smears (Wright-Giemsa): promonocytes have abundant cytoplasm with azurophilic granules that are myeloperoxidase negative, nuclei have delicate folds, nucleoli are inconspicuous
3-6% of AML; affects all ages Mature monocytes or promonocytes predominate in peripheral blood (<80% of monocyte lineage cells are monoblasts, usually <20%) |
Acute monocytic leukemia (M5b)
Micro: leukemic cells are often promonocytes with less basophilic cytoplasm and more azurophilic granules than monoblasts; have folded or cerebriform nuclei with fine chromatin; erythrophagocytosis is common |
|
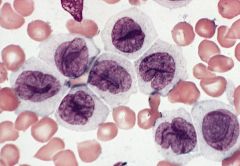
blood smears (Wright-Giemsa): promonocytes have abundant cytoplasm with azurophilic granules that are myeloperoxidase negative, nuclei have delicate folds, nucleoli are inconspicuous
3-6% of AML; affects all ages Mature monocytes or promonocytes predominate in peripheral blood (<80% of monocyte lineage cells are monoblasts, usually <20%) |
Acute monocytic leukemia (M5b)
Micro: leukemic cells are often promonocytes with less basophilic cytoplasm and more azurophilic granules than monoblasts; have folded or cerebriform nuclei with fine chromatin; erythrophagocytosis is common |
|
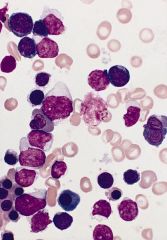
bone marrow smear (Wright-Giemsa): numerous myeloblasts and erythroid precursors at all stages of maturation
|
Acute erythroid leukemia (M6)
Erythroleukemia (M6a) Criteria for diagnosis: 50% of nucleated marrow cells are erythroid lineage, including erythroblasts, 20%+ of nonerythroid cells are myeloblasts; dyserythropoiesis is prominent |
|
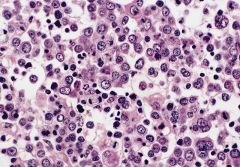
bone marrow biopsy: early to late stage erythroblasts, small megakaryocytes at upper and lower margins, marked reduction in granulocytes
|
Erythroleukemia (M6a)
|
|
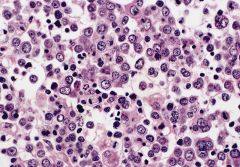
bone marrow biopsy: early to late stage erythroblasts, small megakaryocytes at upper and lower margins, marked reduction in granulocytes
|
Erythroleukemia (M6a)
|
|
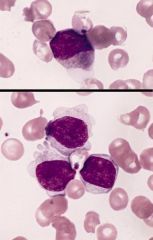
Up to 10% of AML in children (common with Down’s syndrome, see below), 5% of adult AML
Associated with marrow fibrosis due to megakaryoblast secretion of fibrogenic cytokines, which makes marrow aspirate difficult to obtain |
Acute megakaryoblastic leukemia (AMKL, M7)
Criteria for diagnosis: 50%+ blasts of megakaryocyte lineage are present in bone marrow; megakaryocytic lineage based on CD41+ or CD61+ or positive platelet peroxidase reaction on EM |
|
|
- AML with t(8;21)(q22;q22) (AML1/ETO)
- AML with inv(16)(p13q22) or t(16;16)(p13;q22) (CBFβ/MYH11) - Acute promyelocytic leukemia (AML with t(15;17)(q22;q12) (PML/RARα) and variants - AML with 11q23 (MLL) abnormalities |
Acute myeloid leukemias with recurrent genetic abnormalities
|
|
|
Acute myeloid leukemia with multilineage dysplasia
|
Acute myeloid leukemia with multilineage dysplasia
|
|
|
- Alkylating agent related
- Topoisomerase II inhibitor related |
Acute myeloid leukemia and myelodysplastic syndrome, therapy related
|
|
|
- AML minimally differentiated (M0)
- AML without maturation (M1) - AML with maturation (M2) - Acute myelomonocytic leukemia (M4) - Acute monoblastic and monocytic leukemia (M5a and M5b) - Acute erythroid leukemia (M6) - Acute megakaryoblastic leukemia (M7) - Acute basophilic leukemia - Acute panmyelosis with myelofibrosis - Myeloid sarcoma |
Acute myeloid leukemia not otherwise categorized
|
|
|
- Undifferentiated acute leukemia
- Bilineal acute leukemia - Biphenotypic acute leukemia |
Acute leukemia of ambiguous lineage
|