Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
265 Cards in this Set
- Front
- Back
|
What is hemostasis?
|
coagulation that is a physiologic response to injury
|
|
|
What is thrombosis?
|
pathological activation of the coagulation system together with activation and recruitment of platelets
|
|
|
What is the fibrinolytic system?
|
a system that opposes grwoth of the evolving platelet-fibrin thrombus by breaking down or dissolving the fibrin network that is the scaffold of the thormbus.
|
|
|
What are three elements, "Virchow's triad", that predispose a vascular bed to develop thrombosis?
|
alterations in normal blood flow (stasis)
injuries to vascular endothelium alterations in the constitution of blood (hypercoagulability) |
|
|
What is the "workbench" on which coagulation is activated?
|
phospholipid surfaces on platelets and the endothelium
|
|
|
Why is the intrinsic pathway called intrinsic?
|
coagulation components (factors) responsible for forming a thrombus are found within the vascular system (plasma)
|
|
|
Why is the extrinsic pathway called extrinsic?
|
initiates thrombus formation when tissue factor (TF), found in the aventitia around large blood vessels, is exposed to factors within the plasma when the vessel is injured
|
|
|
What is the roll of thrombin (factor II)?
|
central product of coagulation system activation; cleaves fibrinogen to fibrin, which polymerizes into a fibrin network, the scaffold of the clot & activates a number of coagulation factors upstream, thereby increasing the yield of thrombin, and therefore fibrin & activate platelets
|
|
|
What is a zymogen?
|
inactive enzyme precursor
|
|
|
What are the Vitamin K factors?
|
II, VII, IX, and X
|
|
|
What factors are serine proteases?
|
II, VII, IX and X
vitamin k-dependent factors |
|
|
What factors are cofactors?
|
V, VIII, and TF
|
|
|
What is the tenase complex composed of?
|
either Factor IXa + Factor VIIIa or TF:Factor VIIa + Factor IXa
|
|
|
What does the tenase complex do?
|
cleaves factor X to factor Xa
|
|
|
What is in the prothrombin complex?
|
Factor Xa + Factor Va
|
|
|
What does the prothrombin complex do?
|
cleaves Factor II to IIa
|
|
|
Where are K factors synthesized?
|
liver
|
|
|
What role does vitamin K play in forming vitamin K factors?
|
permits the carboxylation of specific glutamate residues in the polypeptide to produce a unique modified form of glutamic acid: gamma carboxyglutamic acid (Gla); the region is a signature of all vitamin K factors
|
|
|
Describe the coagulation cascade: intrinsic, extrinsic, and common pathways.
|
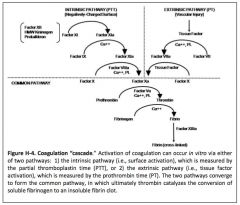
|
|
|
Describe how the intrinsic pathway is activated.
|
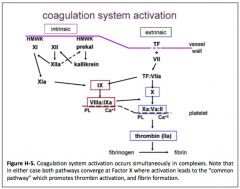
|
|
|
How is is a coagulation factor activated?
|
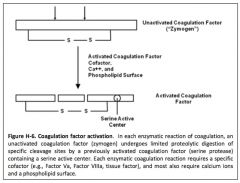
|
|
|
What factors does thrombin "feed back on" and activate?
|
V, VIII, possibly XI
|
|
|
How does thrombin activate the coagulation inhibitor protein C?
|
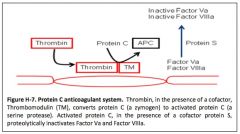
by interacting with its receptor thrombomodulin on endothelial cells.
|
|
|
What is the mechanism of action of tissue factor pathway inhibitor (TFPI)?
|
inhibits Factor VIIa/TF
|
|
|
What is the mechanism of action of antithrombin?
|
binds to active site serine of coagulation of enzymes (thrombin, Xa, IXa)
|
|
|
What pharmaceutical accelarates antithrombin action?
|
heparin
|
|
|
What is the mechanism of action of Protein C?
|
once activated by thrombin/thrombomodulin, activated protein C degrades cofactors Va and VIIIa. Protein S and factor V are also cofactors for this reaction.
|
|
|
What are three regulators of the coagulation system?
|
tissue factor pathway inhibitor (TFPI)
antithrombin protein C |
|
|
How does protein C work?
|
binds to thrombomodulin on endothelial cells, in a step necessary for the activation of PC to APC. Activation takes place when thrombomodulin, stabilized by an endothelial cell protein C receptor (EPCR), receives thrombin and protein C in a complex; APC then binds to its cofactor, protein S. In this form APC dampens the coagulation system and formation of fibrin by proteolytic inactivation of the coagulation factors, factor Va, and factor VIIIa
|
|
|
What is the half life of t-PA? How is it removed from the system?
|
very short 4-5 minutes
inhibition by a circulating inhibitor of plasminogen activation, plasminogen activator inhibitor-1 (PAI-1), as well as clearance by the liver |
|
|
Where is urokinase (u-PA) from?
|
derived from tissue (e.g. kidney) and vascular sources
|
|
|
What is urokinase (u-PA)?
|
physiologic plasminogen activator
|
|
|
Describe the fibrinolytic system.
|
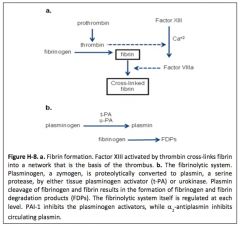
|
|
|
What are two exogenous plasminogen activators (PA) that have been preapred? Where did they come from?
|
streptokinase - streptococci
staphylokinase - staphylococcus |
|
|
What are the regulators of endogenous fibrinolytic system?
|
inhibitors of plasminogen: PAI-1, 2, 3
alpha2-antiplasmin stongly binds any free plasmin withiin circualtion |
|
|
What two synthetic agents with anti-fibrinolytic activity are used clinically? How to they function? Contraindicated in?
|
E-aminocaproic acid
tranexamic acid directly inhibiting plasmin NO in DIC |
|
|
What fragments are the result of degradation of circulating fibrinogen by plasma?
|
X, Y, D, E
|
|
|
What do FDP assays measure?
|
all plasmin degradation products of both fibrinogen and fibrin together
|
|
|
What is the effect of elevated FDP levels on coagulation panels?
|
increaesd FDP levels interfere with normal fibrin polymerization, and thus prolong the thrombin time TT
|
|
|
How can elevated FDP levels effect platelets?
|
They bind platelets and affect platelet function resulting in a prolonged bleeding time.
|
|
|
What does the D-dimer assay measure?
|
DD fragments, which are generated only from plasmin cleaved crosslinked fibrin - marker for ongoing thrombosis
|
|
|
What does activated partial thromboplastin time (aPTT) measure?
|
all of the factors within the intrinsic and common pathways (factors XII, XI, IX, X, VIII, V, prothrombin, and fibrinogen)
|
|
|
What does the prothrombin time measure?
|
factors within the extrinsic and common pathways (factors VII, X, V, prothrombin, and fibrinogen)
|
|
|
What does the thrombin time measure?
|
only the final step in coagulation, the conversion of fibrinogen to fibrin by thrombin
|
|
|
What is a pharmacological inhibitor of coagulation (anticoagulant) which is administered parenteral?
|
Heparin
|
|
|
What is a pharmacological inhibitor of coagulation (anticoagulant) which is administered oral?
|
Warfarin (vitamin K antagonist)
|
|
|
What are two pharmacological inhibitors of thrombin (antithormbin)?
|
antithrombins
anti-factor X inhibitors |
|
|
What pharmaceutical reduces fibrongen levels, and therefore modulates flow?
|
Ancrod
|
|
|
What pharmaceuticals activate plasminogen?
|
recombinant tissue plasminogen activators (rt-PA)
urokinase |
|
|
What is a platelet?
|
an anucleate circulating fragment derived from the cytoplasm of the megakaryocyte in the bone marrow
|
|
|
What are megakaryoctyes derived from?
|
hematopoietic pluri-potenet stem cells
|
|
|
What growth factor influences the development of megakaryocytes and platelet production?
|
thrombopoietin
|
|
|
What is thrombocytopenia?
|
low circulating platelet count
|
|
|
What is thrombocytosis?
|
high platelet count
|
|
|
What is the life span of a platelet?
|
9 - 10 days
|
|
|
What are the two internal membranous systems of the platelet?
|
open canaciluar system - a series of invaginations of the surface membrane that provide route for granule release during activation
dense tubular system - stores calcium that is released during activation and serves as an intracellular second messenger |
|
|
What do dense granules of platelets contain?
|
ADPP
|
|
|
What do alpha granules of platelets contain?
|
multiple proteins, including von Willebrand factor (vWF)- adhesion and fibrinogen- aggreation
|
|
|
Describe the ultrastructure of the platelet.
|
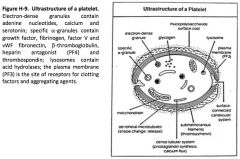
|
|
|
What are the four basic events in the formation of a platelet plug?
|
platelet adhesion
platelet activation , shape change and release reaction platelet aggregation support of local coagulation (fibrin formation and clot retraction) |
|
|
Name four proteins involved in platelet adhesion.
|
vWF, fibronectin, laminin, and thrombospondin
|
|
|
What is the main receptor for adhesion by vWF on platlets? minor receptor?
|
main - Glycoprotein Ib (GP Ib)
minor - glycoprotien IIb/IIIa |
|
|
What are natural inhibitors of platelet aggregation?
|
prostacyclin and nitric oxide
|
|
|
What agonists activate platelets?
|
collagen, ADP, thrombin, epinephrine, thromboxane A2 and calcium
|
|
|
What changes happen in platelets upon activation?
|
cystoskeletal actin filamentous network is altered and platelets become more spherical and extend long pseudopods
|
|
|
What three intracellular processes take place during platelet activation?
|
calcium mobilization, granule secretion and glycoprotein IIb-IIIa
thormboxane A2 formation through archidonic acid and cyclooxygenase cAMp levels are suppressed by agonists resulting in increase Ca++ availability and platelet contraction |
|
|
What substances do activated proteins express?
|
fibrinogen, vWF, ADP, platelet factor 4, and factors V and VIII
|
|
|
When is glycoprotein IIb-IIIa expressed on platelets?
|
only after platelet is activated
|
|
|
What significance does GIIb IIIa have in clot formation?
|
it forms the binding sites for fibrinogen
|
|
|
What is platelet activation?
|
attachment between platelets through fibrinogen molecules acting as bridges, anchored by GPIIb-IIIa
|
|
|
What induces platelet aggregation?
|
ADP, epinephrine, serotonin, thormboxane A2, thrombin & collagen
|
|
|
What is platelet factor 3?
|
negatively charged phospholids that align at the outer surface of the platelet membrane upon activation
|
|
|
On a most basic level, what causes thrombocytopenia?
|
decreased production
increased destruction increased sequestration in the spleen |
|
|
What physical examination signs are seen in thrombocytopenia?
|
hepatosplenomegaly
infection tumor lymphadenopathy |
|
|
What other associated abnormalities might you see on a smear indicative of thrombocytopenia?
|
framgentation hemolysis, abnormal platelet size or presence of abnormal elements
|
|
|
If you have done a through history, PE, and smear, what is the next step in working up thrombocytopenias?
|
bone marrow aspiration and biopsy to determine if marrow megakaryotcytes appear normal in number and to look for other abnormalities such as myelodysplasia or infiltrative diseases
|
|
|
What are the usual manifestations of thrombocytopenia?
|
mucocutaneous bleeding - including petechiae and small ecchymoses, mucous membrane bleeding (epistaxis, gastrointestinal bleeding and menorrhagia)
|
|
|
In patients with younger platelets and thrombocytopenia, what symptom might you get but were not expecting?
|
"hyperfunctional" platelets, which may give less bleeding than expected
|
|
|
In patients with platelet function defects, what symptom might you get but were not expecting?
|
may have more severe bleeding than expected by the platelet count alone
|
|
|
Name three different hereditary thrombocytopenias.
|
May-Hegglin anomaly
Gray platelet syndomre Thrombocytopenia with Absent Radii (TAR) Wiscott Aldrich Syndrome |
|
|
With respect to thrombocytopenias, how should you evaluate a disorder of decreased platelet production?
|
bone marrow examination for the presence of megakaryocytes
|
|
|
What are the causes of decreased platelet production?
|
generalized marrow abnormality (radiation, leukemia, aplastic anemia, folate and vitamin B12 deficiency)
infiltrative marrow processes alcohol, thiazide diuretics and viral infections |
|
|
Why might liver dysfunction lead to decreased platelet production and thrombocytopenia?
|
thrombopoietin is made in the liver and may be significantly decreased in hepatic failure and/or might be multifactorial associated with cirrhosis
|
|
|
What is the status of megakaryocytes in disorders of increased platelet destruction and thrombocytopenia?
|
normal to increased as the marrow tries to increase production in response to the thrombocytopenia
|
|
|
What is auto-immune thrombocytopenic purpura (ITP) caused by?
|
an auto-antibody directed against glycoproteins on the platelet membrane, which are cleared in the spleen, liver and bone marrow
|
|
|
Despite increased numbers of megakaryocytes, why might you still see a decreased platelet production in auto-immune thrombocytopenic purpura (ITP)?
|
probably due to rapid destruction of young platelets in the bone marrow and auto antibody effects on megakaryocytes
|
|
|
In 90% of affected children, what is the clinical course of auto-immune thrombocytopenic purpura (ITP)?
|
is acute and resolves within months
|
|
|
In adults, is auto-immune thrombocytopenic purpura (ITP) acute or chronic?
|
chronic
|
|
|
In adults, how does auto-immune thrombocytopenic purpura (ITP) usually present?
|
occurs for the first time following a viral illness and relapses are frequently preceded by viral illnesses
|
|
|
In adults, is auto-immune thrombocytopenic purpura (ITP) more common in men or women? When is it likely to present in this group?
|
young women
present for the first time or be exacerbated during pregnancy and antibody may cross the placenta, affecting the fetus. If severe, ITP presents with mucosal and cutaneous bleeding. Women may report menorrhagia. |
|
|
What is menorrhagia?
|
abnormally heavy and prolonged menstrual period at regular intervals
|
|
|
How is the diagnosis uto-immune thrombocytopenic purpura (ITP) made?
|
based on normal megakaryoctyes in the bone marrow and exclusion of other causes of platelet destruction such as drugs or DIC; antibody is frequently bound in other conditions, so it is not helpful here.
|
|
|
How do you treat auto-immune thrombocytopenic purpura (ITP)?
|
corticosteroids
if ineffective, splenectomy small portion of patients will require: rituximab, vincristine, danazol, cytotoxic/immunosuppresive agents and intravenous IgG |
|
|
What novel treatment is being used to treat auto-immune thrombocytopenic purpura (ITP)?
|
thrombopoietin mimetic agents
|
|
|
What set of diseases are associated with auto-immune thrombocytopenic purpura (ITP)?
|
HIV, Hepatitis C, systemic lupus and lymphoreticular malignancies (lymphoma, chronic lymphocytic leukemia)
|
|
|
When dealing with thombocytopenia associated with Hepatitis C virus, what might be necessary before antiviral therapy?
|
treatment with TPO mimetic agents
|
|
|
How can immune thrombocytopenia be caused by drugs?
|
A drug-antibody complex bound to the platelet surface is recognized and leads to platelet destruction. Alternatively, some drugs can induce lupus-like syndrome and production of anti-platelet auto-antibodies
|
|
|
What is the treatment for drug-induced immune thrombocytopenia?
|
corticosteroids are NOT effective; removal of the offending drug is most important
|
|
|
What two drugs are the best examples of drug-induced immune thrombocytopenia?
|
quinine and quinidine
|
|
|
How should you treat a patient you suspect has heparin induced thrombocytopenia?
|
Stop all heparin as soon as you suspect; continue antigcoagulation therapy with direct thrombin (DTI) inhibitor, such as hirudin, lepirudin, or aragotroban
|
|
|
What is heparin induced thrombocytopenia associated with?
|
thrombosis due to the formation of platelet plugs
50% morbidity and mortality prolonged anticoagulation with a DTI is then necessary |
|
|
What is post-transufsion purpura (PTP)?
|
seen in patients who lack a common surface antigen; patient develops antibodies to the antigen by exposure through blood transfusion or pregnancy; subsequent transfusions/exposures may cause thormbocytopenia about ONE week later by a mechanism that is not completely clear
|
|
|
How do you treat post-transfusion purpura (PTP)?
|
treatment consists of plasma exchange or intravenous IgG
|
|
|
What is neonatal alloimmune thrombocytopenia (NAITP)?
|
patients develop antibodies to platelet surface antigen after transfusion/pregnancy; these antibodies cross placenta
if inherited antigen from father, severe thrombocytopenia in the fetus and it is at high risk of intracranial bleeding |
|
|
What are two mechanisms for thrombocytopenia in HIV positive patients?
|
decreased platelet production - virus directly affects the marrow with abnormal megakaryoctye growth in culture + disorders that take over bone marrow + antiretroviral therapy
chronic auto-immune thrombocytopenic purpura (ITP) - same as regular ITP |
|
|
What is the treatment for auto-immune thrombocytopenic purpura (ITP) in patients with HIV?
|
responsive to steroids and splenectomy, although may cause further immune supression
intravenous IgG and anti-Rh immuneglobulin can often raise platelet count quickly, Tx with antiretroviral therapy often leads to increaesd platelet count |
|
|
What is the cause of thormbo thrombocytopenic purpura (TTP)?
|
deficiency in the plasma protease, ADAMTS13, which usually cleaves very long multimers of vWF; these multimers aggregate and catch platelets, leading to agglutination at areas of high shear stress = thrombotic microangiopathy, red cell shearing, and possible end organ ischemic damage
|
|
|
What is the major histopathologic finding in thormbo thrombocytopenic purpura (TTP)?
|
platelet thrombi in the microcirculation
|
|
|
What does the blood smear in thormbo thrombocytopenic purpura (TTP) show?
|
schistocytes and decreased numbers of platelets
|
|
|
What is the classic pentad of abnormalities in thormbo thrombocytopenic purpura (TTP)?
|
thormbocytopenia
neurologic abnormalities microangiopathic hemolytic anemia renal abnormalities fever |
|
|
What is the treatment of thormbo thrombocytopenic purpura (TTP)?
|
foremsot plasma exchange therapy although the addition of immunosupressants may be helpful; refractory cases (10%) try splenectomy or rituximab
|
|
|
How does hemolytic uremic syndrome (HUS) typically present?
|
renal function abnormalities which accompany microangiopathic hemolytic anemia and thrombocytopenia
|
|
|
What is hemolytic uremic syndrome (HUS) typically associated with?
|
E. coli O157:H7
can be triggered by drugs such as quinine, cyclosporine and mitomycin C |
|
|
What is the treatment for hemolytic uremic syndrome (HUS)?
|
resolves spontaneously without plama exchange therapy
support dialysis if does remit quickly |
|
|
What about thrombocytopenia in pregnancy?
|
patients do not require therapy and there is not an increased risk of neonatal thrombocytopenia requiring delivery by C-section
neonate might have thrombocytopenia several days following delivery |
|
|
What disease cause hypersplenism and thrnombocytopenia?
|
congestive splenomegaly
infiltrative disease infections or inflammatory responses hyperplastic responses |
|
|
What causes congestive splenomegaly?
|
hepatic cirrhosis, portal vein obstruction, congestive heart failure
|
|
|
What diseases cause infiltrative hypersplenism and thrombocytopenia?
|
Gaucher's disease
other reticuloendothelioses Lymphoma Meyloproliferative diseases |
|
|
What disease cause hyperplastic responses assocaited with hypersplenism and thrombocytopenia?
|
chronic hemolysis
Sickle-hemoglobin C disease Hereditary spherocytosis Autoimmune hemolytic anemia |
|
|
What should management of thrombocytopenic patient be directed at?
|
underlying cause
|
|
|
When should platelet transfusions be given?
|
thrombocytopenia + bleeding
prophylactically in actutely ill patients with platelet counts <10-20,000/microL to prevent bleeding |
|
|
As a general rule, how much will the platelet count rise per unit?
|
5,000 - 10,000/microL
|
|
|
What is the average survival of a transfused platelet?
|
3-5 days
|
|
|
What is seen in the bone marrow with reactive thrombocytosis?
|
increased numbers of normal megakaryocytes
|
|
|
What is the cause of reactive thrombocytosis?
|
iron deficiency
inflammatory diseases and malignancy |
|
|
What is the treatment for reactive thrombocytosis?
|
does not appear to result in any clinical sequelae or increased thrombotic risk
treat underlying illness |
|
|
What is essential thrombocytosis (ET)?
|
myeloproliferative disorder where the major manifestation is thrombocytosis
|
|
|
What does the bone marrow show in essential thrombocytosis (ET)?
|
increased numbers of megakaryocytes, sometimes bizarre in appearance
fibrosis may be seen as well as increased numbers of other hematopoietic precursors |
|
|
What mutation can often (50%) be seen in patients with essential thrombocytosis?
|
JAK2 mutation
|
|
|
What does the blood smear in essential thrombocytosis look like?
|
clumps of platelets, platelets may be extremely large
|
|
|
Why might there be no correlation between platelet count and bleeding in essential thrombocytosis, leading to either prolonged bleeding time or thrombosis?
|
platelets might be abnormal - longer bleeding time
platelets might be hyperreactive - increased thrombosis |
|
|
How should you treat essential thrombocytosis?
|
if a patient has signs or symptoms of either bleeding or thrombosis, the platelet count should be lowered into the normal range with alkylating agents or 32phosphorus
|
|
|
What is anagrelide used to treat? What is its probable mechanism of action?
|
essential thrombocytosis
decreasing platelet "budding" from the megakaryocyte |
|
|
What are some disorders associated with thrombocytosis that might present similar to essential thrombocytosis?
|
myeloproliferative disorders such as polycythemia vera, chronic myelogenous leukemia and myeloid metaplasia
|
|
|
Is there any increased risk of thrombosis or bleeding in post-splenectomy thrombocytosis?
|
no
|
|
|
How will hereditary disorders of platelet function typically present?
|
bleeding manifested will be mucocutaneous - petechiae, ecchymoses, recurrent epistaxis, gastrointestinal hemorrhage, menorrhagia and immediate-type bleeding after trauma and surgical procedures
|
|
|
What are two disorders that cause platelet membrane abnormalities, leading to a hereditary aggregation defect? What is their cause?
|
Bernard-Soulier Disease - Absense of membrane GPIb
Thrombasthenia - absence of membrane GPIIb/IIIa |
|
|
What are two disorders that cause platelet granule abnormalities, producing aggregation defects? What is their cause?
|
Gray Platelet Syndrome - absence of alpha granules
Dense Granule Deficiency - decreased dense ADP |
|
|
What disorder cause platelet plasma factor deficiency, producing aggregation defects? What is its cause?
|
von Willebrand Disease - vWF deficiency w/ decreased factor VII levels
|
|
|
What is von Willebrand factor?
|
synthesized by endothelial cells and megakaryocytes and stored in alpha-granules of the platelets and in endothelial cells; important in platelet adhesion and as a carrier as a carrier protein for factor VII
|
|
|
What reaction is abnormal in vWF disease?
|
in-vitro platelet aggregation response to the antibiotic risocetin
|
|
|
What drugs inhibit platelet function through affecting prostanoid synthesis?
|
aspirin
corticosteroids Others (idomethacin, phenylbutazone, ibuprofen, fenoprofen, naproxen, sulfinpyrazone, furosemide, vitamin E) |
|
|
What drugs inhibit platelet function through activating adenylate cyclase?
|
prostanoids (prostacyclin, PGE1, PGD2)
Others (isoprenaline, adenosine) |
|
|
What drugs inhibit platelet function through inhibition of phosphodiesterase?
|
Pyrimidopyrimidines (dipyridamole)
Methyl xanthines (caffeine, theophylline, aminophylline, papaverine) |
|
|
What drugs inhibit platelet function as microbial agents?
|
Penicillins and cephalothins (carbenicillin, penicillin G,
ticarcillin, ampicillin, cephalosporin) Others (nitrofurantoin, ristocetin, hydroxychloroquine) |
|
|
What drugs inhibit platelet function by membrane stabilization?
|
Local anesthetics (procaine, xylocaine)
Antihistamines (diphenhydramine, promethazine) Tricyclic antidepressants (imipramine, nortriptyline) |
|
|
What drugs inhibit platelet function by blocking sympathetics?
|
Alpha-antagonists (phentolamine)
Beta-antagonists (propanolol) |
|
|
What drugs inhibit platelet function by inhibiting GP IIb/IIIa?
|
Abciximab (Rheopro®) anti GPIIb/IIIa monoclonal
antibody Eptifibatide (Integrilin®)-heptapeptide GPIIb/IIIa binder Tirofiban (Aggrastat®)- non-peptide GPIIb/IIIa binder |
|
|
What conditions can cause activation of platelet membrane and release of storage pool material?
|
distrubed platelet vessel interactions:
cardio-pulmonary bypass valvular heart disease cavernous hemangioma DIC thermal injury leads to circulation of "exhausted platelets" |
|
|
What systemic disorders can affect platelet function?
|
renal failure, liver disease, amyloidosis
multiple myeloma |
|
|
What does a increase in mean platelet volume (MPV) mean?
|
may be seen in some cases of myelodysplasia or with increased platelet production resulting in circulation of larger, younger platelets
|
|
|
What is hemophilia?
|
group of hereditary genetic disorders that impair the body's ability to control blood clotting or coagulation
|
|
|
What factor is deficient in hemophila A?
|
factor VIII
|
|
|
What factor is deficient in hemophila B?
|
factor IX
|
|
|
What is the inheritance pattern of hemophila?
|
X-linked recessive
|
|
|
Which disease is more common, hemophila A or hemophila B?
|
hemophila A is 3-4 times more common
|
|
|
How can you diagnose hemophilia?
|
show isolated prolonged PTT
differentiating A vs. B requires specific actor assays (VIII and IX, respectively) |
|
|
What is the characteristic clinical features of hemophila?
|
deep tissue (e.g. joint, muscle) bleeding is characteristic of both type of hemophilia
|
|
|
What are the factor VIII or IX level and bleeding manifestations in persons with severe hemophilia?
|
less than 1% normal of factor
spontaneous bleeding occurs at rest or with daily activities |
|
|
What are the factor VII or IX level and bleeding manifestations in persons with mild hemophilia?
|
1-5% of normal factor
spontaneous bleeding is less common, bleeding occurs with trauma or surgery |
|
|
What are the factor VII or IX level and bleeding manifestations in persons with mild hemophilia?
|
6-30% of normal factor
bleeding only following trauma or surgery |
|
|
Why must we treat severe hemophila promptly?
|
may develop chronic synovitis and arthritis, airway obstruction via hematoma, muscle atrophy
|
|
|
In women who inherit the abnormal or hemophilic factor VII gene, what is their approximate factor VIII levels?
|
50%
|
|
|
How many cases of hemophilia are spontaneous (de novo) mutations?
|
50%
|
|
|
What is the treatment for hemophilia?
|
prompt replacement of the deficient factor to hemostatic levels during episodes of acute or potential bleeding - target level is 30 - 100% of factor
|
|
|
How can you manage mild hemophilia A?
|
with desmopressin or DDAVP, a vassopressin analog that results in release of stored factor VIII from endothelial cells
not successful until stores have been repleted (2-3 days); not useful in severe hemophiliacs |
|
|
How long after surgery must factor levels be kept high in hemophiliacs?
|
14 days
|
|
|
What adjunctive agent is useful in hemophilia (A or B) following dental extractions?
|
e-aminocaproic acid - and antifibrinolytic agent
|
|
|
What are the most serious treatment complications in treatment of hemophilia?
|
transmission of HIV and Hep viruses
screening has made this much better recombinant factors are becoming available |
|
|
What are the complications in treatment of hemophilia?
|
virus contraction
development of factor VIII or IX alloantibody inhibitors |
|
|
What is the usual inheritance patter of vWF disease?
|
autosomal dominant
|
|
|
How do you diagnose vWF disease?
|
prolonged bleeding time - due to platelet adhesion defect
prolonged PTT - due to low circulating factor VIII levels "Ristocetin cofactor assay" - platelet aggregation in presence of ristocetin is abnormal vWF multimer analysis distinguishes types |
|
|
What is Type I vWF disease?
|
quantitative deficiency of vWF
|
|
|
What is Type II vWF disease?
|
qualitative deficiency of the largest vWF multimers
|
|
|
What are the clinical features of vWF disease?
|
relatively mild bleeding tendency that is predominately mucocutaneous
temporally variable - unpredictable after surgery or trauma |
|
|
What is the inheritance pattern of Type III vWF disease? What are its clinical features?
|
Autosomal recessive
severe bleeding resembling severe factor VIII deficiency or hemophilia A |
|
|
When do we expect increases in vWF?
|
during pregnancy, hyperthyroidism, renal failure, and the stress of exercise, inflmmation, trauma or surgery
|
|
|
How do we treat vWF disease?
|
infusions of desmopressin (DDAVP) - check responsiveness
vWF concentration do not use cryoperipitate because of risk of viruses |
|
|
Besides hemophilia A and B, what other factor deficiencies are reported? One interesting thing about them?
|
XI - Ashkenazi Jews
XII - markedly long PTT but asymptomatic XIII - delayed bleeding + poor wound healing, normal coags |
|
|
How do you diagnose a vitamin K deficiency?
|
PT is most sensitive, PTT may also be prolonged
|
|
|
What pharmaceutical is a vitamin K antagonist?
|
warfarin (coumadin) competitively inhibits coagulation factor carboxylation
|
|
|
What can cause vitamin K deficiencies?
|
generalized fat malabsorption because vitamin K is a fat soluble vitamin and absorption is dependent on pancreatic lipases and bile
|
|
|
Where can humans obtain vitamin K?
|
leafy green vegetables
sysnthesis by gastrointestinal bacterial flora |
|
|
How do we treat vitamin K deficiency?
|
if immediate risk, rapid reversal via fresh frozen plasma (FFP) containing all the vitamin K dependent factors may be necessary
parenteral vitamin K may take up to 24 hours (prophylaxis is given to all newborns) |
|
|
How can liver disease cause an acquired coagulation disorder?
|
liver is site of synthesis of all coagulation factors except vWF and VIII
site of carboxylation of vitamin K dependent factors associated with production of abnormal fibrinogen molecule (dysfibrinogenemia) - prolonged TT major site of clearance of activated factors, as well as of breakdown products (FDPs) develop thrombocytopenia - cirrhosis & portal hypertension = splenic sequestration |
|
|
How can you diagnose liver disease as a cause of a coagulation disorder?
|
PT can be prolonged
advanced disease - PT and PTT prolonged |
|
|
What causes the thrombocytopenia found in liver disease?
|
folate deficiency
toxic EtOH effects increase in splenic pooling decrease in thrombopoietin production |
|
|
What causes the prolonged bleeding time in liver disease?
|
increase in fibrin(ogen) degradation products
|
|
|
What causes the prolonged PT and PTT in liver disease?
|
decrease in vitamin K dependent carboxylation
decrease in factor synthesis |
|
|
What causes the prolonged thrombin time in liver disease?
|
dysfibrinogenemia
decreased clearance of fibrin(ogen) degradation products |
|
|
What causes the low fibrinogen level seen in liver disease?
|
decreased synthesis
|
|
|
How should you manage the coagulopathies associated with liver disease?
|
correction of any underlying anatomic or vascular defect
supportive blood components whole blood if bleeding if PT or PTT > 1.5 x normal - fresh frozen plasma but patients with ascities have reduced half-lives |
|
|
What is the pathophysiology of DIC?
|
results from the release of tissue factor, enzymes, or some other procoagulant substance from damaged or abnormal tissue, that ultimately results in activation, consumption and/or degradation of coagulation factors, coagulation inhibitors, and platelets
|
|
|
How do you diagnose DIC?
|
constellation of findings - lab & clinic
thymbocytopenia + hypofibrinogenemia prolongation of PT, PTT, TT FDP or D-dimer elevation |
|
|
When do you see DIC?
|
bacterial sepsis, severe tissue injury, hypotension and or anoxia, obstetrical emergenies, and vascular disorders that involve extensive endothelial disruption
chronic can be seen in malignancies |
|
|
How do you treat DIC?
|
treat underlying cause
|
|
|
How do you tell the difference between the presence of a coagulation inhibitor and a coagulation factor deficiency?
|
"1:1 mix" - if the prolonged screening test corrects to normal when patient plasma is diluted 1:1 with normal plasma, deficiency
if the plasma fails to correct upon addition of normal plasma, inhibitor |
|
|
What are three examples of coagulation inhibitors?
|
lupus anticoagulant, heparin, factor VII inhibitor
|
|
|
What are usually specific coagulation inhibitors?
|
IgG antibodies - can develop as:
alloantibodies to factor replacement autoantibodies to factor |
|
|
How do you treat patients with coagulation inhibitor antibodies?
|
immunosuppressive therapy
acute bleeding episodes - high dosese of human factor VIII concentrate, procine factor VIII concentrates, or factor VIII bypass produces such as crude factor IX concentrate or recombinant factor VIIa |
|
|
Paradoxically, what are lupus anticoagulants associated with?
|
thormbotic tendency as well as spontenous abortions
anticoagulant therapy may be indicated |
|
|
What are thrombi that form in high flow systems composed of? What are these called?
|
platelet aggregates which are held together by stands of fibrin
white thrombi |
|
|
What are thrombi that form in slow flow or stasis composed of? What are these called?
|
rich in red blood cells with large amount of interspersed fibring
red thrombi |
|
|
What causes vascular endothelium abnormalities?
|
injury
immune complexes viruses hemodynamic stress (hypertension) tobacco ulceration of cholesterol plaques enzymes released from platelets and WBC in inflammatory states prosthetic surfaces |
|
|
What should be included in the history of a patient with thrombosis?
|
age at first event
risk factors, incl. pregnancy or hormone therapy vessels involved total number and frequency of events family history? previous or current antithrombotic therapy general health |
|
|
How can you prevent venous stasis in a hospitalized patient?
|
early ambulation after surgery and external leg compression devices. Low dose heparin (both standard and low molecular weight can be used prophylactically and is highly effective with only minimal increased risk of bleeding. Coumadin is also effective when started prior to surgery.
|
|
|
What are the clinical findings in deep vein thrombosis (DVT)?
|
pain, tenderness, swelling and discoloration
A positive Homan sign tender "cord" that can be palpated along the posterior calf or inner thigh that corresponds to the thrombosed vessel |
|
|
What is post-phlebitic syndrome?
|
chronic venostasis as a result of recurrent venous thrombosis of the legs
|
|
|
What causes the pleuritic chest pain in pulmonary embolisms?
|
inflammatio of the pleura overlying areas of pulmonary infarction or atelectasis results in pleuritic chest pain, characterized by sharp pain that increases with inspiration
|
|
|
What signs might be found on the cardiovascular exam with a pulmonary embolism?
|
right ventricular lift
loud pulmonary second sound - increased pulmonary vascular resistance jugular venous pressure elevation distended neck veins present gallop rhythm due to right ventricular failure |
|
|
How do you diagnose DVT?
|
contrast venogram is reference but invasive so use:
B-mode ultrasonography doppler flow studies D-dimer has strong negative predictive value ventalation/perfusion (V/Q) lung scan (85% if high probability) |
|
|
How do you manage a patient with venous thrombosis or pulmonary embolism?
|
Immediate heparinization is first! - results in an immediate inhibition of screeing clotting tests - watch PTT
Oral antigcoagulation via Warfarn after fully anticoagulated on heparin |
|
|
How is heparin usually administered initially (drug regime)?
|
intravenous bolus in an amount sufficient to result in immediate anticoagulation. An initial loading dose is about 75 units per kg of body weight. This is followed by a continous intravenous infusion in dosage sufficient to achieve a PTT two to three times the normal range. A hematocrit and platelet count should be obtained prior to beginning therapy and monitored 1-2 days for bleeding/thormbocytopenia.
|
|
|
Is there a risk of developing heparin induced thrombocytopenia (HIT) with low molecular weight heparin (LMW)?
|
Less likely to cause than heparin but may cross react with heparin antibodies already formed and should not be administered to a patient with HIT
|
|
|
How does warfarin/coumarin work?
|
chemically similar to vitamin K; stops final carboxylation step in vitamin K-dependent factor synthesis
|
|
|
What factor is most effected by warfarin?
|
Factor VII - shortest half life
|
|
|
What test do you follow to monitor warfarin therapy?
|
PT
|
|
|
What is the goal INR for persons with DVT or PE?
|
2-3
|
|
|
What kinds of medications should you use caution with when prescribing warfarin?
|
warfarin is highly albumin bound and may be displaced by other medications
|
|
|
When is warfarin used?
|
three to six months to prevent recurrence in patients with venous thrombosis
used chronically in some with recurrent thrombotic disease |
|
|
What are the complications of anticoagulation therapy?
|
bleeding - incidence is higher in patients on warfarin and monitored less frequency
any new or painful symptom on anticoagulated patient requires evaluation for potential bleeding complication |
|
|
How do you reverse heparin?
|
protamine sulfate
|
|
|
How do you reverse warfarin?
|
requires infusion of fresh frozen plasma to the patient to raise the vitam K-dependent factor levels immediately
give Vitamin K also but will take several days to reverse warafarin |
|
|
Besides heparin induced thrombocytopenia (HIT), what other side effect is associated with heparin?
|
osteoporosis
|
|
|
What is a mural thrombosis?
|
in contact with the endocardial lining of a cardiac chamber or, if not occlusive, with a wall of a large blood vessel
|
|
|
How do you diagnose an arterial thrombosis?
|
doppler flow studies but specific diagnosis will require contrast angiography
|
|
|
How do you manage arterial thrombosis and thormboemolic disease?
|
remove mechanically if big enough
thrombolytic therapy |
|
|
What are thrombolytic agents?
|
plasminogen activators - promote clot lysis by converting plasminogen to the fibrinolytic enzyme, plasmin
|
|
|
What is the time frame needed to effectively administer thrombolytic agents after acute coronary artery occlusion?
|
w/in 6 hours of onset of symptoms
|
|
|
What are the indications for using thrombolytic therapy in treatment of acute occlusion of peripheral arteries?
|
acute embolic occlusion of a majory artery
patients with contraindications to surgical revascularization treatment of peripheral arterial occlusions that are not amenable to surgical correction |
|
|
What is the major risk for thrombolytic therapy? What patients is this therapy counter indicated in?
|
hemorrhage
recent neurosurgery, head trauma, or CNS hemorrhage intracranial neoplasm or aneurysm stroke w/in 6 months active or recent internal bleeding uncontrolled hypertension surgery or trauma in prior 2 weeks pregnancy |
|
|
What are methods of prevention of arterial thrombosis or thromboembolism?
|
aspirin - anti-platelet agent
heparin in unstable angina heparin and Rehopro (Ab against GP IIb/IIIa) following angioplasty of coronary vessel Warfarin in atrial fibrillation, heart valves |
|
|
What clues can a patient provide that point to a thrombotic disorder?
|
history of recurrent thrombosis, family history of thormbosis or sudden death, thrombosis at a young age (<45), and thrombosis at locations other than the deep veins of the legs
|
|
|
What patients without thrombosis, should be screened for thrombophilia?
|
relatives with thrombophilia
SLE patients for lupus inhibitor women with poor obstetric history or recurrent spontaneous abortions |
|
|
What patients with thrombosis should be screened for thrombophilia?
|
single event in patient < 40 y/o w/risk factors
recurrent thormboses postive family history single unusual event or with serious sequeale |
|
|
How is activated protein C (APC) resistance or Factor V Leiden corrected?
|
by factor V and is associated with a specific factor V polymorphism that makes Factor V resistant to the action of protein C
|
|
|
What is the inheritance pattern of protein C and protein S deficiencies?
|
autosomal dominant
|
|
|
What are deficiencies in protein C and protein S associated with clinically? How is this treated?
|
neonatal purpura fulminans - a rare disorder that can rapidly pogress to death if not treated with replacement therapy (fresh frozen plasma)
|
|
|
How are patients with deficiencies in protein C and S routinely treated?
|
heparin
long term warfarin for recurrent thrombosis |
|
|
What is warfarin-induced necrosis? When does it happen?
|
rapid decrease in protein C (or S) causes necrosis upon initiation of warfarin therapy
|
|
|
What is the consequence of the newly found prothrombin mutation?
|
Prothrombin G20210A has a C to A transition at a single nucleotide encoding prothrombin - associated with 3-5 fold increased risk of venous thrombosis; prothrombin levels are freq. elevated
|
|
|
What is the inheritance pattern of antithrombin III deficiency?
|
autosomal dominant; heterozygotes display intermediate levels of antithrombin activity associated with a thrombotic tendency
|
|
|
What is the usual role of antithrombin III?
|
inactivates thrombin and other serine proteases (XIIa, XIa, Xa, IXa) by irrevestible binding to the active serine site of the protease
|
|
|
How can I detect an Anthrombin III activity abnormaility?
|
ATIII heparin cofactor assay is useful in both quantitative and qualitative abnormalities
|
|
|
How are acute thrombotic events usually managed in antithrombin III deficient patients?
|
heparin thearpy but may require ATIII concentrate or plasma to provide ATIII substrate
|
|
|
What are the two mechanisms by which nephrotic syndome is associated with increased risk of thrombosis?
|
decreased levels of ATIII and protein S due to loss in the urine
|
|
|
When does a hemolytic transfusion reaction occur?
|
destruction may occur if the patient has antibodies directed toward donor red cell antigen or, more rarely, if transfused plasma contains high-titer antibodies to the patient's red cells
|
|
|
What are the signs and symptoms of acute intravascular hemolytic reaction?
|
fever, chills, anxiety, hypotension, dyspnea, bronchospasm, chest pain, back pain, a buring sensation in the vein used for the administration of the transfusion, urticaria, nausea, and vomiting
|
|
|
In patients under anesthesia, what might be the only signs of intravascular hemolytic reaction?
|
hypotension and diffuse hemorrhage due to dIC
|
|
|
What are the complications of intravascular hemolysis?
|
DIC and acute renal failure
|
|
|
Why does DIC occur in intravascular hemolysis?
|
thromboplastin effect of released red cell strome as well as the effect of immune complexes
free hemoglobin may also play a role in binding nitric oxide |
|
|
After you recognize a hemolytic transfusion reaction, what do you do?
|
immediately discontinue transufsion
immediately examine patients plasma for presence of free hemoglobin - will look red attempts should be made to maintain urine flow by diuretics and sufficient intravenous fluids send samples to blood bank to determine cause |
|
|
When do delayed transfusion reaction occur?
|
in patients who have been previously sensitized
no antibody is detectable abut there is an unexplained drop in patients hematocrit 3-10 days later w/fever, hemoglobinuria and hyperbilirubinemia |
|
|
What is a febrile transfusion reaction usually due to?
|
antibodies to white cells
|
|
|
What are the signs/symptoms of a febrile transfusion reaction?
|
fever and/or chills beginning 30 minutes to two hours after the start of the transfusion
|
|
|
How do you control a febrile transfusion reaction?
|
with aspirin or acetominophen
corticosteroids or meperidien may be required for more severe reactions transfusion need not be discontinued if the symptoms are mild |
|
|
What are the signs and symptoms of allergic transfusion reactions?
|
uicaria, hypotension, bronchospasm, or anaphylaxis
|
|
|
What causes allergic transfusion reactions?
|
antibodies to plasma proteins
|
|
|
How do you treat mild cases of allergic transfusion reactions?
|
antihistamines
slow transfusion |
|
|
How do you treat severe cases of allergic transfusion reactions?
|
corticosteroids or epinephrine
life threatening allergic reactions may require cell washing |
|
|
What is transfusion-related lung injury (TRALI) due to?
|
interaction of leukocyte antibodies (usually transfused) and leukocytes - there is leukoagglutination in the pulmonary microvasculature and subsequent pulmonary damage
|
|
|
Describe the signs and symptoms of TRALI?
|
appear within 30 minutes to 6 hours of transfusion, include dyspnea, fever, bilateral pulmonary infiltrates, and hypoxia
|
|
|
What infections can be transmitted via blood transfusions?
|
Hepatitis
HIV CMV |
|
|
What is transfusion-associated graft versus host disease (TAGVHD)?
|
a fatal disorder that may occur if transfused immunocompetent lymphocytes survive in the recipient, a situation most likely to occur if the patient is immunoincompetent or if the donor and recipient are HLA closely matched
|
|
|
How can you prevent transfusion-assocaited graft versus host disease (TAFVHD)?
|
irradiate blood products for intrauterine transfusion, neonatal exhchange transfusion, transfusions form relatives or HLA matched donors, and for patients with immunodeficiency syndromes, lymphoproliferative diseases, leukemias, or stem cell transplantation
|