Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
54 Cards in this Set
- Front
- Back
|
What are the three types of point mutations?
|
Silent: altered DNA codes for the same amino acid without changing the phenotypic effect.
Missense: altered DNA codes for a different amino acid, which changes the phenotypic effect. Nonsense: altered DNA codes for a stop codon that causes premature termination of protein synthesis. |
|
|
Define and name some examples of a trinucleotide repeat disorder.
|
Errors in DNA replication that causes amplification of a sequence of three nucleotides. Anticipation is associated (increasing severity in generations). Examples: Huntington's, Fragile X, Friedreich's ataxia, myotonic dystrophy.
|
|
|
If the carrier rate of CF is 1/25, what is the prevalence of CF in the population?
|
1/25 * 1/25 * 1/4
|
|
|
What is the inheritance of the following protein disorders and what is the defective protein:
1) Hereditary angioedema 2) Hereditary spherocytosis 3) Neurofibromatosis 4) Hemophilia A 5) Duchenne's muscular dystrophy |
1) AD: C1 esterase inhibitor
2) AD: ankyrin 3) AD: neurofibromin 4) X-linked rec: Factor VIII 5) X-linked rec: dystrophin |
|
|
What is the inheritance and pathogenesis of phenylketonuria?
|
PKU is an AR genetic disorder characterized by a deficiency of phenylalanine hydroxylase, causing a decrease in tyrosine. Phenylalanine is metabolized into neurotoxic phenylketones and acids that produce mental retardation and urine with a musty odor.
|
|
|
Homogentisate oxidase is an enzyme in the pathway of tyrosine entering the citric acid cycle as fumarate. What disease is due to the absence of homogentisate oxidase and what are the clinical features?
|
Alkaptonuria: degenerative arthritis in spine, hip and knee; black urine when oxidized by light; sclera darken (ochronosis).
|
|
|
What enzyme is deficient in McArdle's disease? Hint: glycogen accumulates.
|
Muscle phosphorylase. Clinical features: muscle fatigue; no increase in lactic acid with exercise; myoglobinuria with strenuous exercise.
|
|
|
Pompe's is an autosomal recessive disorder characterized by a deficiency of a lysosomal enzyme. What enzyme is this? Hint: glycogen accumulates. In the infant form, what is the most common cause of death?
|
Pompe's is a lack of alpha-1,4-glucosidase which is needed for the breakdown of glycogen. Mental retardation, hypotonia, and cardiomegaly leading to death at 2 year of age in the infant form.
|
|
|
Maple syrup urine disease is due a deficiency in the enzyme needed to breakdown isoleucine, leucine, and valine. What is this enzyme? What is the inheritance of this disease?
|
Enzyme: branched chain alpha-ketoacid dehydrogenase.
Autosomal recessive. Clinical features: mental retardation, seizures, feeding problems, sweet-smelling urine. |
|
|
What is the inheritance of these disorders and which is the most common:
Wilson's 21-hydroxylase deficiency Thalassesmia Hemochromatosis |
All are autosomal recessive. Hemochromatosis is the most common AR disorder.
|
|
|
Hurler's and Hunter's disease are very similar, but Hunter's is a less severe form. What is the genetic inheritance of these two disorders and what enzyme's are deficient?
|
Hurler's: AR; deficiency of alpha-L-iduronidase leading to lyosomal accumulation of dermatan sulfate and heparan sulfate; clinical features include retardation, coarse facial features, hepatosplenomegaly, corneal clouding, coronary artery disease.
Hunter's: X-linked; deficiency of iduronate sulfatase with the same build of proteins. |
|
|
What is the genetic inheritance of Marfan's syndrome?
|
Autosomal dominant disorder resulting in the production of weak elastic tissue due to a defect in synthesizing fibrillin. CF's: MVP, aortic dissection, eunuchoid proportions.
|
|
|
What is the inheritance of the following disorders:
Huntington's Osteogenesis imperfecta Achondroplasia Tuberous sclerosis |
All are autosomal dominant. Remember that Huntington's also shows anticipation because it is a trinucleotide repeat disorder.
|
|
|
Male presents with mental retardation, speech delay and appears hyperactive (as well as attention deficit). His physical features include macroorchidism, a prominent jaw, and large ears. You confirm this man has the most common X-link disorder. What is it?
|
Fragile X syndrome, which is also a trinucleotide repeat disorder (CGG). Fragile X syndrome is the second leading cause of inherited mental retardation.
|
|
|
What percent of female carriers of Fragile X syndrome may develop mental retardation?
|
50%.
|
|
|
How is the enzyme HGPRT (hypoxanthine-guanine phosphoribosyl transferase) utilized in the body?
|
HGPRT i sinvolved in the salvage of the purines hypoxanthine (converted to IMP) and guanine (converted GMP). HGPRT utilizes PRPP as the source of ribose 5-phosphate to form IMP and GMP.
|
|
|
What enzyme is deficient in Lesch-Nyhan syndrome and what is the inheritance?
|
HGPRT (hypoxanthine-guanine phsphoribosyltransferase) is used in the salvage of purines hypoxanthine (to IMP) and guanine (to GMP) using PRPP. L-N is an X-linked disorder characterized bymental retardation, hyperuricemia, and self-mutilation.
|
|
|
What is the inheritance in Bruton's agammaglobulinemia?
What is the inheritance of the variant of SCID characterized by adenosine deaminase deficiency? |
Bruton's: X-linked recessive.
SCID: autosomal recessive. |
|
|
A patient has glomerulonephritis, sensorineural hearing loss and some ocular defects. You work out that he has a family history of all these symptoms and it effects males and females equally. What is the disease?
|
Alport's syndrome is an X-linked dominant disorder caused by defective type IV collagen.
|
|
|
What is the inheritance of the following disorders and how would you describe the bones of a patient with one of these disorders:
1) Vit D resistant Rickets 2) Osteogenesis imperfecta |
1) X-link dominant: softening of bones (bowed legs).
2) AD: brittle bone disease. |
|
|
What is the Lyon hypothesis?
|
In females, one of the two X chromosomes is randomly inactivated. Inactivation occurs in the embryonic period. The inactivated X chromosome is called a Barr body. Normal females have one Barr body, normal males have none.
|
|
|
How does triploidy happen?
|
Triploidy: 69 chromosomes. (1) Failure of meiosis in a germ cell (fertilization of a diploid egg by a haploid sperm), (2) dispermy (two sperm that fertilize one egg).
|
|
|
Mixoploidy is a condition where a person has two or more genetically different cell populations. What is this condition called when the cell populations arise from a single zygote? Different zygotes?
|
Mosaicism is when the genetically different cell populations arise from a single zygote. It usually occurs via nondisjunction of chromosomes during mitotic division in the early embryonic period. Chimerism is different zygotes.
|
|
|
What is aneuploidy?
|
Addition of one chromosome - trisomy.
Loss of one chromosome - monosomy. |
|
|
What is the consequence of nondisjunction in meiosis one?
|
Production of a gamate with 22 chromosomes and one with 24.
|
|
|
What is a Robertsonian translocation? What is the consequence of a Robertsonian translocation between 14q21q?
|
This is a balanced translocation between two acrocentric chromosomes. The long arms (q) combine, while the short arms (p) combine and degenerate. Carriers are clinically normal. The issue in this occurs when the carriers produce gametes by meiosis and reproduce. Depending on how the chromosomes segregate during the meiosis, conception can produce trisomy 21 (live), trisomy 14 (miscarriage), monosomy 14 or 21 (miscarriage), normal complement, or a t(14q21q) carrier.
|
|
|
What is the genetic defect in Cri Du Chat?
|
Deletion: loss of a portion of chromosome. Cri du chat syndrome is a loss of the short arm of chromosome 5. CF: mental retardation, cat-like cry, ventricular septal defect, microcephaly.
|
|
|
What percent of Down syndrome is caused by nondisjunction?
|
95%. 4% - Robertsonian. 1% mosaicism).
|
|
|
What are Down patients at greater risk of?
|
AVSDs, Hirschsprung's, duodenal atresia, AML (< 3 years old), ALL (> 3 years old), Alzheimers by 35, sterility in all males, females have 50% of having a child with Down.
|
|
|
What is the genetic defect in Edwards syndrome?
|
Trisomy 18 which is characterized by mental retardation, clenched hands with overlapping fingers, VSD, and early death.
|
|
|
What is the genetic defect in Patau's syndrome?
|
Trisomy 13 which is characterized by mental retardation, cleft lip and palate, polydactyly, VSD, cystic kidneys, and early death.
|
|
|
What is the chromosomal make up of Turner's syndrome? What is the cardinal feature?
|
Monosomy X (45,X). This condition is only found in females and the cardinal feature is short stature (normal GH and IGF-1).
|
|
|
A patient presents with amenorrhea and delayed sexual maturity. The female patient has normal intelligence. Further history reveals as a newborn her hands and feet were swollen and she had a webbed neck. What lab values could help you confirm your suspicions? What karyotype could this patient have?
|
The patient has turner's syndrome, which is the most common genetic cause of primary amenorrhea. Labs: decreased estradiol, increased FSH and LH, no Barr body.
The two possible karyotypes: (1) Nondisjunction 45, X (60%), (2) mosaicism 45,X/46,XX (40%). |
|
|
What ovarian changes are seen in Turner's patients?
|
Streak gonads: ovaries replaced by fibrous stroma. Ovaries devoid of oocytes by 2 years of age. Increased risk for developing ovarian dysgerminoma.
|
|
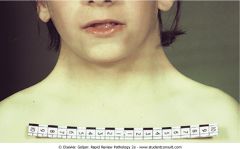
What kidney abnormality may this patient have?
|
Turner's syndrome (45,X). Horseshoe kidney.
|
|
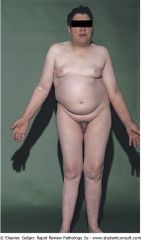
What is the testosterone level, FSH, and LH levels of this patient?
|
Klinefelter's syndrome: decrease testosterone and inhibin, increased LH and FSH.
|
|
|
What are the clinical features of Klinefelter's syndrome?
|
XXY; nondisjunction.
Female 2nd sex characteristics at puberty: peristent gynecomastia, soft skin, female hair distribution. Delayed sexual maturation (hypogonadism): testicular atrophy, fibrous seminiferous tubules, loss of Sertoli, Leydig cell hyperplasia. Eunuchoid habitus. One Barr body. |
|
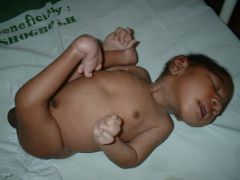
The patient has rocker-bottom feet overlapping fingers. Diagnosis?
|
Trisomy 18 - Edwards syndrome.
|
|
|
Mitochondrial diseases exhibit maternal inheritance, where affected mothers transmit the disease to all their children. What tissues are most susceptible to a mitochondrial disease?
|
mtDNA-encoded proteins are associated with electron transport and ATP synthesis, therefore, tissues with a high oxygen demand are most affected by mitochondrial dysfunction.
|
|
|
What type of inheritance is displayed by hereditary optic neuropathy?
|
Mitochondrial disease (if mother is affected, all children inherit the disease). Caused by defect in complex I of ETC (NADH dehydrogenase) which cuases progressive loss of central vision and blindness due to the degeneration of the optic nerve.
|
|
|
What mitochondrial DNA disorder is characterized by ophthalmoplegia (degeneration of the motor nerves of the eye), pigment degeneration of the retina, and complete heart block?
|
Kearns-Sayre syndrome.
|
|
|
A biopsy reveals ragged red fibers in a skeletal muscle biopsy which shows abnormal shaped mitochondria. The patient from which the biopsy was taken is suffering from myoclonus, seizures, and ataxia. Diagnosis?
|
Myoclonic epilepsy.
|
|
|
What is genomic imprinting?
|
The differential expression of alleles depending on whether the allele is on the paternal or maternal chromosome. The expression of the paternal (or maternal) chromosome depends on the DNA methylation patterns. Increased methylation of DNA = silenced genes.
|
|
|
Prader-Willi gene is normally inactivated on the maternal chromosome 15 and activated on the paternal chromosome 15. How does Prader-Willi syndrome occur?
|
A microdeletion (15q11-13) derived from the father.
|
|
|
A child presents with hyperphagia (insatiable appetite), hypogonadism, hypotonia, obesity, short stature, small hands, and violent behaviour. A karyotyping shows an abnormality on chromsome 15. Diangosis?
|
Prader-Willi syndrome. A microdeletion on the paternal chromosome 15q results in this disorder. This highlight the principle of genomic imprinting (normal child - maternal chr15 is inactivated via methylation, paternal chr15 is activated).
|
|
|
What is the clinical consequence of a microdeletion in the q arm of chromosome 15 derived from the mother?
|
Angelman syndrome (Happy puppet syndrome): mental retardation, wide-based gait, inappropriate laughter.
|
|
|
In normal sex differentiation, what does an absence of a Y chromosome result in?
|
Wolffian (mesonephric) duct structure undergo apoptosis. Germinal tissue differentiates into ovaries.
If present: Mullerian inhibitory factor causes mullerian tissue to undergo apoptosis. MIF is synthesized in the Sertoli cells. Testosterone causes the Wolffian duct structures to develop (epididymis, seminal vesicles, vas deferens). |
|
|
1) What is a true hermaphrodite?
2) What is a pseudohermaphrodite? |
1) Fetus has both male/female goonads. Karyotype is usual 46, XX.
2) Phenotype and genotype do not match. Male: XY with testes but female phenotype. Female: XX with ovaries but male phenotype. |
|
|
What is the most common cause of male pseudohermaphroditism?
|
Testicular feminization. XR disorder with a deficiency of androgen receptors. Fetal DHT and testosterone are unable to function without receptor.
|
|
|
During what week of embryogenesis is the fetus most susceptible to disturbances in the morphogenesis of an organ - malformations?
|
4th and 5th weeks.
|
|
|
During what week of embryogenesis is the fetus most susceptible to extrinsic disturbances in fetal development - deformations?
|
Occur between the 9th week and term after fetal organs have developed. Most often due to restricted movement in uterine cavity.
|
|
|
What is the consequence to the fetus of oligohydramnios?
|
Oligohydramnios from decreased production of fetal urine (renal agenesis) restricts fetal movement in the uterine cavity. Newborns have flat facial features (Potter's facies) and underdevelopment of the chest wall and clubfeet.
|
|
|
Define:
1) Agenesis 2) Aplasia 3) Hypoplasia 4) Atresia |
1) Complete absence of an organ resulting form absens of the primordial tissue (anlage).
2) Anlage is present but never develops. 3) Anlage develops incompletely, but histologically normal. 4) Incomplete formation of a lumen. |
|
|
What is the congenital anomaly that can occur in a newborn whose mother has SLE?
|
If the mother with SLE has anti-Ro antibodies the newborn may develop congenital heart block.
|