![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
31 Cards in this Set
- Front
- Back
- 3rd side (hint)
|
Acute lymphoblastic leukemia/lymphoma (ALL) |

Associated with Down syndrome after age 5. TDT = DNA polymerase. Must give CNBS and testis prophylaxis.
ALL can be divided into B-ALL (CD 10, CD 19, CD 20) and T-ALL (CD2-CD8, no CD10, thymic mass as teenager)
B-ALL: t(12;21) has a good prognosis (kids mostly) t (9;22) poor prognosis |
HIGHLIGHTS •Clinical history plus most signs and symptoms are nonspecific and reflect bone marrow dysfunction. They include fatigue, dyspnea, dizziness, bleeding, easy bruising, and recurrent infections. •Physical examination may reveal pallor, ecchymoses, lymphadenopathy, or hepatosplenomegaly. There may also be evidence of tissue infiltration such as bone pain, testicular enlargement, and cranial nerve symptoms and signs. •Definitive diagnosis requires bone marrow biopsy. Presence of blast cells in greater than 25% of the bone marrow cells confirms the diagnosis of acute lymphocytic leukemia (ALL). Initial peripheral blood smear may show leukemic lymphoblasts. •Treatment uses multiagent dose-intense chemotherapy regimens in induction, consolidation, and maintenance phases. Early stem cell transplantation considered in selected patient population. •Despite the aggressive management methods, less than 30% of adults with ALL can be cured. Long-term adverse effects of treatment include heart failure, sterility, secondary malignancies, and neurotoxicity. ETIOLOGY •Genetics, including genetic disorders: trisomy 21, Klinefelter syndrome, and inherited diseases with excessive chromosomal fragility such as Fanconi anemia, Bloom syndrome, and ataxia-telangiectasia •Environmental factors: include exposure to atomic bomb explosions, radiation, smoking, use of hair dyes, and employment in electrical occupations. PATHOPHYSIOLOGY •Molecular abnormalities seen in ALL can be classified according to the functional consequence of oncogenic mutation. Activation of the ABL protein kinase via rearrangement with the BCR gene is an example of a mutation that results in a proliferative advantage. The most common cytogenetic abnormality in adult ALL results from chromosomal translocation t(9;22)(q34;q11), the Philadelphia chromosome. [1] [3] [17] [19] [20] [21] [22] •Other gene rearrangements may result in loss or gain of function mutations involving transcription factors that play a role in hematopoietic development. An example of such gene rearrangement is the t(12;21)(p13;q22) chromosomal translocation that juxtaposes the TEL genes. [1] [3] •Other mechanisms of cancer formation involve loss or inactivation of tumor-suppressor genes via deletions and gene rearrangements. Examples of such mechanisms involve p16(INK4A) and p53. [1] [3] [18] •FLT3 and NOTCH1 have been identified as genes mutated in MLL/hyperdiploid and T-ALL, respectively. [23] CREBBP mutations are seen in 18% of relapsed ALL and may confer resistance to therapy. [24] PAX5 gene is mutated in up to 30% of pediatric patients with ALL. [25] [26] IKZF1 mutations may be a predictor of relapse. [27] PHF6 mutations are seen in 38% of adult T-ALL samples. [28] CDKN2A mutations are seen in 42% of cases of T-ALL. [29] Much of this data has yet to lead to risk stratification or alternative therapies. PRESENTATION •Key Diagnostic Factors •Factor Frequency •lymphadenopathy common •hepatosplenomegaly common •pallor, ecchymoses, or petechiae common •fever common •fatigue, dizziness, palpitations, and dyspnea common •epistaxis, menorrhagia common • DIAGNOSIS •1st Tests To Order •Test Result •CBC with differential anemia, leukocytosis, leukopenia, and/or thrombocytopenia •serum electrolytes elevated serum levels of calcium, potassium, phosphorus, uric acid, and lactic acid may be seen •peripheral blood smear leukemic lymphoblasts •renal function urea may be normal or elevated •liver function liver enzymes may be normal or elevated •lactic dehydrogenase may be elevated •coagulation profile results are variable •bone marrow aspiration and trephine biopsy bone marrow hypercellularity and infiltration by lymphoblasts •immunophenotyping (on bone marrow, or peripheral blood if cell count is raised) ALL blasts express surface antigens and molecular markers that help to identify their specific lineage and demonstrate aberrant myeloid antigens and biphenotypic or bilineage variants •thiopurine methyltransferase (TPMT) phenotype varying expression in population •cytogenetics variable •BCR/ABL molecular studies presence of the Philadelphia chromosome • SERUM ELECTROLYTES •The degree of uric acid elevation reflects the extent of tumor burden. •Hypercalcemia may be caused by bony infiltration or ectopic release of a parathormone-like substance. •Phosphorus may be elevated due to ineffective leukopoiesis or as a result of chemotherapy-induced tumor lysis. •Hyperkalemia may also occur as a result of extensive leukemic cell lysis. TREATMENT •Standard induction therapy for ALL includes prednisone (or dexamethasone), vincristine, anthracyclines, and/or L-asparaginase. Other drugs, such as cyclophosphamide, cytarabine, mercaptopurine, or intrathecal methotrexate may be added as part of early intensification protocols. A cautious cell reduction phase is recommended for patients with a large leukemic burden, determined by WBC more than 25,000/microliter. This may be achieved by a combination of corticosteroids and vincristine or cyclophosphamide. In addition, leukapheresis is indicated in cases with symptomatic leukostasis prior to initiation of therapy. Patients should be closely monitored for tumor lysis syndrome after the start of therapy. [49] [50] •All patients receive CNS prophylaxis, as it forms part of the induction therapy regimen. Prophylactic treatments of CNS leukemia may result in acute or chronic neurotoxicity presenting as pyrexia, arachnoiditis, leukoencephalopathy, and milder subclinical CNS dysfunctions. COMPLICATIONS •Panacytopenia •Tumor lysis syndrome •Infertility
|
|
|
Small lymphocytic lymphoma (SLL)/ chronic lymphocytic leukemia (CLL) |

With SLL, involvement in lymph nodes causes generalized lymphadenopathy.
In both, autoimmune hemolytic anemia occurs because of self-reactive immunoglobulins. |
Common Vignette 1 A 62-year-old man presents at the primary care physician's office for an annual physical. He denies any complaints such as fever or chills, weight loss, or fatigue. Of note, his blood tests show an elevated WBC count. The WBCs are predominantly lymphocytes, with a differential of 80% lymphocytes and an absolute lymphocyte count of 75,000/microliter. Common Vignette 2 A 60-year-old man presents with swollen lymph nodes in the cervical and inguinal region that have been present for 2 months and are gradually increasing in size. The lymphadenopathy is painless and has not responded to a course of antibiotics prescribed by the primary care physician. The patient denies any recent history of infection, fever, or chills. Other Presentations The majority of patients present with lymphadenopathy (87%). Around half present with splenomegaly, and occasionally some will present with hepatomegaly (14%). One quarter of these patients have no symptoms and an elevated absolute lymphocyte count on a routine blood test. The typical B symptoms of lymphoma (unintentional weight loss, fever with a temperature >101°F, night sweats, and fatigue) can occur in CLL but are much less frequent (10%). [2] /// HIGHLIGHTS Hematologic malignancy that occurs with increasing age. Presents with asymptomatic lymphadenopathy or with absolute lymphocytosis as an incidental finding on routine CBC. Diagnosed by CBC, with differential, blood smear showing smudge cells, and flow cytometry. Variable clinical course. Clinical and laboratory criteria are used to determine treatment approach and prognosis. Treatment options include a conservative (watch and wait) approach or chemotherapy. Median survival is around 2 to 3 years in high-risk patients and 25 years in good-risk patients. The cause of death depends on age and comorbidity. EPI a. Primarily occurs in individuals > 60 years old • Median age for CLL is 65 years old. b. Most common overall leukemia in Western countries c. Most common cause of generalized lymphadenopathy in the same age bracket ETIOLOGY Unknown PATHOPHYSIOLOGY • Neoplastic disorder of virgin B cells (B cells that cannot differentiate into plasma cells) PRESENTATION •Generalized painless lymphadenopathy (Fig. 13-11A ) •Metastatic sites similar to those of AML •Increased incidence of immune hemolytic anemia • Warm (IgG) or cold (IgM) immune hemolytic anemia may occur Key Diagnostic Factors Factor Frequency shortness of breath and fatigue common lymphadenopathy common splenomegaly common Other Diagnostic Factors Factor Frequency B symptoms uncommon recurrent infections uncommon petechiae uncommon DIAGNOSIS • Laboratory findings •a. Peripheral WBC count ranges from 15,000 to 200,000 cells/mm3 (see Fig. 13-11B ) •b. Lymphoblasts are < 10% of the total lymphocyte count. •c. Neutropenia •d. Numerous “smudge” cells (fragile leukemic cells) •e. Normocytic anemia (50% of cases) and thrombocytopenia (40% of cases) •f. Bone marrow findings •(1) Usually completely replaced by neoplastic B cells •(2) Lymphoblasts account for < 10% of the lymphoid cells that are present. •g. Hypogammaglobulinemia •• Neoplastic B cells do not develop into plasma cells. •5. Five-year survival rate is 75%. •1st Tests To Order •Test Result •WBC count with differential elevated with absolute lymphocytosis >5000/microliter •blood film smudge cells present •hemoglobin Hb <110mg/L (11.0 g/deciliter) •platelet count platelets <100 x10^3/microliter •flow cytometry CD5, CD19, and CD23 positive •Other Tests to Consider •Test Result •fluorescent in situ hybridization variable; may show cytogenetic abnormalities such as del(13q14), del(11q), or del(17p) •Coombs test positive test indicates autoimmune hemolytic anemia •immunoglobulin levels hypogammaglobulinemia •bone marrow aspirate and core biopsy variable; may show marrow infiltration by leukemic cells, reduction in hematopoetic precursor compartment •CT scan variable; may show hepatosplenomegaly; retroperitoneal or mediastinal adenopathy • TREATMENT •Patients with good physical performance and favorable cytogenetics should receive fludarabine, cyclophosphamide, and rituximab (FCR) combination chemotherapy. [7] [8] [9] [26] •Patients with unfavorable cytogenetics (del(17p) or p53 mutations) respond poorly to fludarabine. In the absence of specific clinical protocols, alemtuzumab as monotherapy, or in combination with other agents in the context of a clinical trial, should be offered. [8] Such patients should be considered for an allogeneic stem cell transplant after initial response.
|
|
|
Hairy cell leukemia |

Cladribine works because adenosine accumulates to toxic levels in neoplastic B cells. |
Common Vignette A middle-aged man presents with complaints of acute onset of abdominal discomfort and fullness, as well as weight loss. The patient also complains of shortness of breath and fatigue. Physical examination reveals pallor and splenomegaly. Other Presentations Other presentations of HCL may include anemia, neutropenia, or thrombocytopenia. /// HIGHLIGHTS • ETIOLOGY Unknown, although woodworking and Epstein-Barr have been mentioned PATHOPHYSIOLOGY •Splenomegaly can be attributed to diffused infiltration of red pulp cords and sinuses accompanied by atrophy of pulp cords, which results in sequestration, marginalization, and destruction of healthy blood cells inside the spleen. Bone marrow failure and splenomegaly cause pancytopenia in HCL. Accumulation of hairy cells and reticulin fibrosis in the bone marrow and the detrimental effects of dysregulated cytokine production cause bone marrow failure. •Note that splenomegaly occurs in most chronic leukemias •Hairy cells are clonal late B cells that express a range of B-cell markers. PRESENTATION •Key Diagnostic Factors •Factor Frequency •abdominal fullness or discomfort common •splenomegaly common •Other Diagnostic Factors •Factor Frequency •weakness and fatigue common •pallor and petechiae common •fever common •recurrent infections common •hepatomegaly common • DIAGNOSIS •CBC: pancytopenia •Peripheral blood smear shows hariy cells •Immunophenotyping (huge list) TREATMENT •Treatment with purine analogs is the standard of care and is used as a first-line therapy for patients with HCL. It has replaced splenectomy as initial treatment of choice except in rare cases of splenic rupture or massive splenomegaly. •The risk for infections in patients with neutropenia and monocytopenia is high, and an active stance regarding the workup and treatment of infections before starting treatment with purine analogs is recommended. [11] •Owing to ease of administration, a single cycle of cladribine Evidence may be preferred over pentostatin Evidence by some practitioners, particularly when the subcutaneous route of administration is used. COMPLICATIONS •Patients with HCL are at increased risk for secondary malignancies. Approximately 30% of patients with long-term survival will develop secondary tumors
|
|
|
Acute myelogenous leukemia (AML) |

MPO+
Patho: Retinoic acid receptor distrupted, leading to increased promyelocytes. ATRA binds to the receptor, leating to maturation of blasts and treatment/cure.
Can get AML from preexisting dysplasia (alkylating agents, radiotherapy). Can lead to myelodysplastic syndrome, which lead to AML.
t(15;17)
Promyelocytes have Auer rods.
Increased risk of DIC |
Common Vignette A 58-year-old man presents to his primary care physician with increasing tiredness, accompanied by bruising on his legs. He also complains of aching bones. He has no previous illnesses. On examination, he is pyrexial and pale, has bony tenderness over the sternum and tibia, and petechiae on his legs. There are no palpable lymph nodes. He has crepitations at the left base. The liver and spleen are not palpable. Other Presentations Other presentations of AML include hepatosplenomegaly or lymphadenopathy. Less commonly, tumors may present with gingival enlargement, skin chloromas, and soft tissue or meningeal leukemic infiltration. Coagulopathies are reasonably common with AML, and DIC is common with acute promyelocytic leukemia (APML). An elevated WBC count >100,000/microliter is found in approximately 10% of patients, predisposing them to complications such as tumor lysis syndrome, CNS involvement, and leukostasis. [3] These are medical emergencies and require immediate treatment. /// HIGHLIGHTS •A life-threatening disorder that predominately occurs in older adults. •Many subtypes exist, of which acute promyelocytic leukemia merits specific management. •Characteristically abnormal blasts are present in the peripheral blood, and normal hematopoiesis is reduced. Definitive diagnosis requires bone marrow biopsy. Presence of blast cells in >20% of the bone marrow cells confirms the diagnosis. •Cytogenetic abnormalities are prognostically important and affect patient management. •Most patients are treated with chemotherapy induction, consolidation, and maintenance regimens. Hematopoietic stem cell transplantation may also be used in select patients. •It is important to rapidly identify acute promyelocytic leukemia, as associated coagulopathy may be life threatening. •Although most patients achieve complete remission, a high incidence of relapse leads to poor overall survival. ETIOLOGY •However, environmental exposure to radiation, benzene, and treatment with chemotherapy drugs are risk factors for developing AML. [7] [8] [9] Smoking is the most common exposure to benzene. Abnormalities of chromosome 5 and 7 are increased in AML due to radiation, benzene, and alkylating chemotherapy. •Cytogenetic abnormalities in AML fall into either unbalanced deletions or losses (5q-/-5, 7q- or -7 or t8), balanced translocations (frequently involving MLL at 11q23, AML1 at 21q22, RAR-alpha at 17q21, and CBF-beta at 16q22), or normal karyotype. PATHOPHYSIOLOGY •In AML, the accumulation of blasts unable to differentiate into mature neutrophils, RBCs, or platelets results in bone marrow failure, which is the commonest cause of death. As there is little correlation between blast percentage and cytopenias, secretion of inhibitory substances such as chemokines, rather than physical replacement of normal marrow by the myeloid blasts, is thought to lead to suppression of normal hematopoiesis. [15] Neutropenia predisposes to severe infections by endogenous aerobic gram-positive and gram-negative bacteria, and Candida and Aspergillus species. [16] Increasing neutrophil counts are often the best predictors of response. Infiltration of the lungs or CNS may occur, usually when the WBC count is >50,000/microliter, and blasts are monocytic or express CD56 antigen on their surface. [17] PRESENTATION •Key Diagnostic Factors •Factor Frequency •pallor common •ecchymoses or petechiae (from thrombocytopenia) common •Other Diagnostic Factors •Factor Frequency •fatigue common •dizziness common •palpitations common •dyspnea common •infections or fever common •lymphadenopathy common •hepatosplenomegaly common •mucosal bleeding common •skin or testicular mass uncommon • DIAGNOSIS •1st Tests To Order •Test Result •CBC with differential anemia, macrocytosis, leukocytosis, neutropenia, and thrombocytopenia •peripheral blood smear blasts on blood film, presence of Auer rods •coagulation panel (If all tests are abnormal (e.g., with prolonged PT and activated PTT, decreased fibrinogen, and elevated D-dimer), DIC should be suspected.) •serum electrolytes may be normal; serum levels of calcium, potassium, phosphorus, uric acid, and lactic acid may be elevated; serum calcium may be decreased •renal function variable; BUN may be elevated •LFTs may be normal or elevated •serum lactic dehydrogenase may be elevated •Other Tests to Consider •Test Result •bone marrow biopsy or aspiration bone marrow hypercellularity and infiltration by blasts; blasts >20%, Auer rods, morphologic description of type of blast •immunophenotyping and molecular studies blasts express surface antigens and molecular markers that help to identify their specific lineage •lumbar puncture variable; malignant cells •HLA antigen typing variable •chest x-ray may show evidence of consolidation, pulmonary infiltrates, cardiomegaly •echocardiogram variable •multigated acquisition scan variable • TREATMENT •Complicated
|
|
|
Chronic myelogenous leukemia (CML) |

LAP -
Predominant cells are granulocytes.
Basophils are characteristically increased
Splenomegaly common; acute leukemia
CML can progress to AML (2/3) or ALL (1/3) |
Common Vignette 1 A 54-year-old man presents to his primary care physician with a 2-month history of fever, malaise, and weight loss. He also reports frequent epistaxis, abdominal fullness, and early satiety. On exam, he is found to have splenomegaly. Common Vignette 2 A 50-year-old man presents to his primary care physician for a routine physical. He is asymptomatic at the time of the visit and the physical exam is normal. Routine baseline bloods showed elevated WBC and platelet counts. Other Presentations Less commonly, patients may present with acute gouty arthritis due to excess production of uric acid from extensive cell turnover, tenderness of the sternal area due to bone marrow over-expansion, blurring of vision due to retinal hemorrhages from high white blood cell count; priapism, and splenic pain due to infarction. [4] /// HIGHLIGHTS •Malignant clonal disorder of hematopoietic stem cells. •At presentation, one third of patients may be asymptomatic; if present, symptoms typically include malaise, fever, weight loss, abdominal discomfort, and night sweats. •Splenomegaly is the most common physical finding; all patients will have elevated WBC count. •Presence of Philadelphia chromosome and /or molecular demonstration of the BCR-ABL transcript confirms diagnosis. •Treatment with a tyrosine kinase inhibitor (e.g., imatinib, dasatinib, or nilotinib), which inhibits the BCR-ABL tyrosine kinase, gives long-term remission without significant adverse events in most patients. • ETIOLOGY •The only known risk is exposure to ionizing radiation. No other environmental toxins or hereditary features have been found so far. PATHOPHYSIOLOGY •The BCR gene on chromosome 22 is fused to the ABL gene originating from the distal portion of chromosome 9, resulting in a BCR-ABL fusion oncogene. PRESENTATION •Key Diagnostic Factors •Factor Frequency •splenomegaly common •shortness of breath uncommon •LUQ discomfort or fullness uncommon •epistaxis uncommon •arthralgia uncommon •sternal tenderness uncommon •Other Diagnostic Factors •Factor Frequency •weight loss common •excessive sweating common •fever uncommon •pallor uncommon •bruising uncommon •retinal hemorrhages uncommon • DIAGNOSIS •1st Tests To Order •Test Result •CBC elevated WBC count; anemia; normal platelet count, thrombocytosis (chronic or accelerated phases), or thrombocytopenia (accelerated or blast crisis) •complete metabolic profile may have elevated potassium, LDH, or uric acid •peripheral blood smear mature or maturing myeloid cells, elevated basophils and eosinophils •bone marrow biopsy granulocytic hyperplasia •cytogenetics Philadelphia chromosome positive t(9,22) •quantitative reverse transcription PCR (qRT-PCR) including breakpoint analysis detection of BCR-ABL fusion •fluorescent in situ hybridization (FISH) t(9,22) positive • TREATMENT •Standard initial treatment is imatinib. COMPLICATIONS •Complication Likelihood Timeframe •pancytopenia (view full topic) high short term •tyrosine kinase inhibitor-related myelosuppression medium variable •tyrosine kinase inhibitor-related muscle cramps medium variable •tyrosine kinase inhibitor-related rash medium variable •tyrosine kinase inhibitor-related QT prolongation medium variable •
|
|
|
Chromosomal translocations and Down syndrome associations |

|
Down syndrome
Age < 5 Acute Megak. Leukemia
Age >5 ALL |
|
|
Langerhans cell histiocytosis |

|

|
|
|
Chronic Myeloproliferative Disorders |

|

|
|
|
Polycythemia table |

|
|
|
|
Leukemoid reaction |

|
1. Leukemoid reaction a. Definition—absolute leukocyte count usually > 25,000 to 30,000 cells/mm3 • May involve neutrophils, lymphocytes, or eosinophils b. Normal bone marrow response to cytokines released by cells (e.g., lymphocytes, stromal cells, macrophages) to infection (most common) or trauma c. Examples include: (1) Neutrophilic leukocytosis associated with perforated appendicitis (2) Lymphocytosis associated with whooping cough (3) Eosinophilia associated with cutaneous larva migrans d. Pathogenesis • Normal albeit exaggerated response to an infection |
|
|
Leukoerythroblastosis (leukoerythroblastic reaction) |

|
13-1: Leukoerythroblastic reaction. The solid arrow shows a teardrop RBC. Immature myeloid cells are also present in a nucleated RBC (interrupted arrow). |
|
|
Neutrophilic leukocytosis |

|
|
|
|
Causes and patho of neutropenia |
Causes (1) Aplastic anemia (2) Immune destruction • Examples—systemic lupus erythematosus (SLE), and paroxysmal nocturnal hemoglobinuria (3) Septic shock (4) Drugs • Penicillin, cephalosporins, quinidine, and sulfonamides (5) Tick-borne diseases • Ehrlichiosis, anaplasmosis, and babesiosis (6) Viral infections • Hepatitis, infectious mononucleosis (7) Bacterial infections • Typhoid fever, brucellosis, and tuberculosis (8) Fungal infections • Systemic fungal infections (e.g., histoplasmosis) (9) Ionizing radiation |
(9) Ionizing radiation c. Pathogenesis (1) Decreased production • Aplastic anemia, ionizing radiation, and drugs (2) Increased destruction • Destruction by complement, macrophages, and antibodies (3) Activation of neutrophil adhesion molecules (a) Increase the number of neutrophils adhering to endothelium (b) Example—endotoxins |
|
|
Iron deficiency anemia |
|
|
|
Pathogenesis of white cell malignancies |
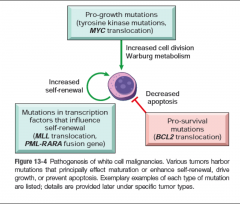
|
|
|
|
|
|
|
|
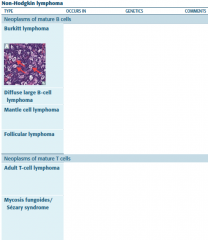
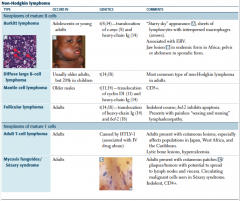
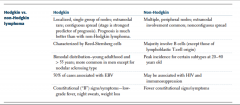
HL vs NHL |
|
|
|

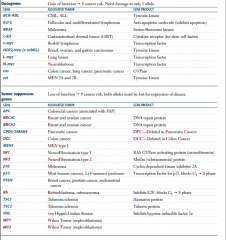
Tumor suppressor or oncogene? |
|
|
|
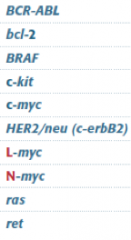
Tumor suppressor or oncogene? |
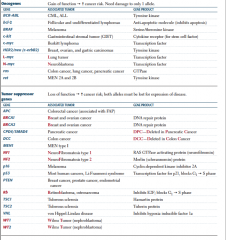
|
|
|
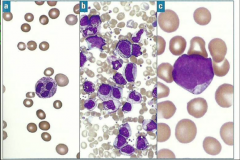
Which type of myeloid disorder?
Blood |
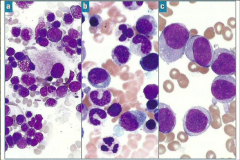
Which type of myeloid disorder?
Bone marrow |
A, B, C
MDS, MPN, AML
|
|

|

|
|
|

|
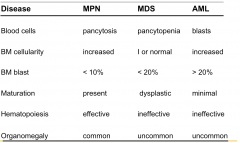
|
|
|
|
Distinguishing AML from ALL |
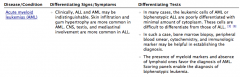
AML:
Age 65 years
MPO+
t(15;17)
Responsibe to ATRA
Auer Rods |

ALL:
Age<15 years, with two smaller peaks at 35 years and 80 years
tdt+
More likely in CNS and testes
t(12;21)
B-ALL (CD10, 19, 20)
T-ALL (CD2-8, no CD10); thymic mass |
|
|
Distinguishing AML and MDS |

|

|
|
|
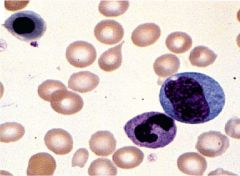
Identify this peripheral blood smear |
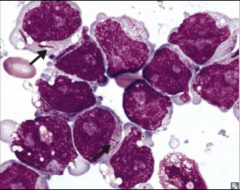
|
AML |
|
|
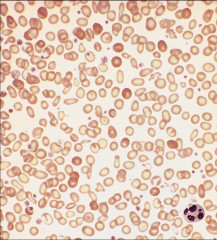
Identify this peripheral blood smear |
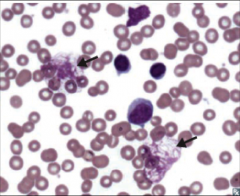
|
CLL |
|
|
Identify this peripheral blood smear |
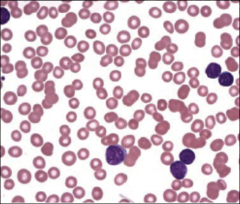
|
ALL |
|
|
Identify this peripheral blood smear |
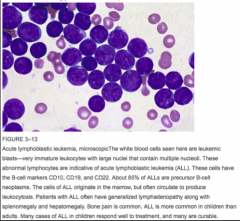
|
|
|
|
Identify this peripheral blood smear |
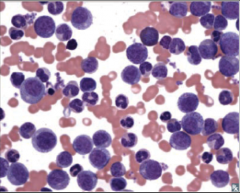
|
CML |
|
|
Identify this peripheral blood smear |
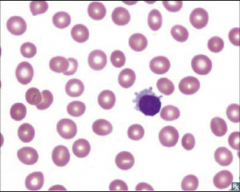
|
Hairy cell leukemia |
|
|
Identify this peripheral blood smear |
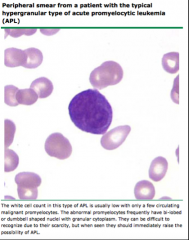
|
|