Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
64 Cards in this Set
- Front
- Back
|
Haemophilia |
Haemophilia is a group of inherited blood discorders of blood coagulation.
*Results in insufficient generation of thrombin from Factor 10a and Factor 8a complex through the intrinsic pathway. 1. Haemophilia A -represents 80% of all haemophilia -deficiency in Factor 8 -40% inversion of gene on Chromosome 10 60% deletion, insertion and point mutations 1:5000 male incidence 2. Haemophilia B -Christmas disease -congenital deficiency in Factor 9 -point mutation and deletions -1:25,000 male incidence Pathophysiology -genetic -acquired haemophilia : development of auto-antibodies -idiopathic >50 yo -peripartum -drug reaction (eg penicillin) -malignancies Titres Severe : <1% normal factor (<0.01 IU/mL) Moderate : normal factor (0.01-0.05 IU/mL) Mild: >5% but less than 40% normal factor (>0.05 to <0.40 IU/mL) Morbidity -Haemarrthragia most common : painful and leads to long-term inflammation and deterioration of the joint -resulting in permanent deformities, misalignment, loss of mobility, and extremities of unequal lengths. Viral complications -HIV -Rates of seroconversion were more than 75% for severe disease, 46% for moderate disease, and 25% for mild disease. -In severe haemophilia B, seroconversion was observed at a rate of 46%. -More than 50% of patients with haemophilia were infected with HIV by 1983. Life-threatening haemorrhage -Intracranial haemorrhage -2-8% risk -1/3 deaths due to ICH -Other life-threatening haemorrhages include soft-tissue haemorrhages that obstruct the airway or damage internal organs. Medical Management -Various Factor VIII and Factor IX concentrates are now available to treat haemophilia A and haemophilia B. -Reductions in infectious complications and improved purity are the main advantages of these concentrates. -During production, a specific viral-inactivation stage, either solvent-detergent treatment or liquid-phase heat treatment, is implemented to inactivate viruses, such as hepatitis B virus, hepatitis C virus, and HIV. -However, the transmission of nonenveloped viruses (eg, parvovirus and hepatitis A virus) and poorly characterized agents (eg, prions) is still a problem. Recombinant Factor VIII and Factor IX are now commercially available. They have lowered the risk of viral contamination. Analgesia -Pain medications are used for acute bleeding or chronic arthritis. Safe analgesics include paracetamol, oxycodone, and morphine (avoid all aspirin products). The treatment of patients with inhibitors of FVIII is difficult. Porcine Factor VIII, which has low cross-reactivity with human factor VIII antibody, has been used. Factor VIII inhibitor-bypassing agents (FEIBA), including Factor IX complex, activated prothrombin complex concentrate (aPCC), and activated Factor VII has also been used. Plasmapheresis, IVIG, or immunosuppressive therapy with cyclophosphamide and prednisolone, have showed some success in achieving long-term control. Rituximab with prednisone plus or minus the addition of mycophenolate mofetil may be used when standard therapy has failed. Anaesthetic Management of Patients with Haemophilia -Intramuscular pre medication should be avoided. -Vascular access does not cause excessive bleeding, but should be placed with care, central venous lines should ideally be placed used ultrasound. -After induction of anaesthesia, extra care should be taken in manipulation or intubation of the airway as it can cause submucosal haemorrhages, which can become a life threatening condition. Nasal intubation should be avoided, as it can prove traumatic and bleeding from the site can lead to aspiration. Care should be taken during positioning of the extremities and pressure points should be padded to prevent intramuscular haematomas or haemarthrosis. Post operatively, analgesics such as NSAIDs should not be given as it can predispose patients to gastrointestinal haemorrhage. Patient-controlled analgesia is a safe and effective alternative to intramuscular injections. All patients should be admitted, even for minor procedures and optimal management should be discussed with the local haematologists. |
|
|
Von Willebrand disease (VWD)
|
Von Willebrand disease (vWD)
-Family of bleeding disorders caused by an abnormality of the von Willebrand factor (vWF) -Most common hereditary bleeding disorder. -Prevalence of 1% Von Willebrand Factor -Large multimeric glycoprotein, carrier protein for F8 -Primary function (platelet adhesion and aggregation) : attaches to glycoprotein 1b on platelet and damaged subendothelium -Secondary (F8 carrier) : protects F8 from degradation and delivers it to the site of inury. Classification of vWD Type 1 vWD -partial quantitative decrease of qualitatively normal vWF and FVIII -autosomal dominant -mild symptoms Type2 vWD -15-20%; autosomal dominant or recessive -5 know subtypes : 2A, 2B, 2C, 2M, 2N -qualitative reduction in function : normal levels of vWF, but the protein is structurally abnormal -associated thrombocytopenia due to abnormal spontaneous platelet binding, worsened by pregnancy, surgery and DDAVP Type 3 vWD -most severe form -autosomal recessive -no detectable vWF antigen -no response to DDAVP Causes Inherited -defect of short arm of chromosome 12 Acquired : o Wilms tumour o Congenital heart disease o Systemic lupus erythematosus o Angiodysplasia o Seizure disorders treated with valproic acid o Hypothyroidism Treatment General measures -consult haemotologist -surgical technique, tourniquet -avoid hypothermia, hypercarbia, acidosis, hypertension -avoid NSAIDS and other anticoagulants Specific -Aim for vWF between 50-100% of normal titres -measure before and after initiation of treatment Type 1 -DDAVP 0.3ug/kg in 30ml NS over 30min Type 2/3 -give F8 concentrates with preserved vWF -FFP (contains F8) and cryo (contains F9) should be only be given in emergencies when F8/vWF unavailable due to risk of viral transmission and factor inhibition generation |
|
|
Guidelines for Jehovah’s Witnesses
|
add
|
|
|
Bleeding patient. What is relative contraindication to Prothrombinex?
A. History of HITS B. Von Willebrands C. Haemophilia B D. Warfarin overdose ?? or - Overdose vit K (not warfarin) E. Renal failure |
ANSWER A
DESCRIPTION Each pack contains one vial of lyophilised high-purity human factor IX (500 IU), factor II (500 IU), factor X (500 IU), antithrombin (25 IU) and heparin (200 IU), one ampoule of Water for Injection BP and a filter needle. INDICATIONS Prothrombinex is indicated only as directed by the Consultant Haematologist. For the prophylaxis and treatment of bleeding in patients with single or multiple congenital deficiencies of factor II or X and in patients with single or multiple acquired prothrombin complex factor deficiency requiring partial or complete reversal (eg. Reversal of Warfarin anticoagulant therapy) Note: Prothrombinex should NOT be used for prophylaxis or treatment of haemorrhage in patients with Haemophilia B. CONTRAINDICATIONS Prothrombinex should NOT be used in patients showing clinical or laboratory signs of thrombosis or DIC. Prothrombinex should NOT be used for prophylaxis or treatment of haemorrhage in patients with Haemophilia B. DOSE AND ADMINISTRATION Dosage and administration should be discussed with the Consultant Haematologist. Dosage varies from 20-30 IU/kg for minor haemorrhage up to 50 IU/kg for moderate to severe haemorrhage. Exact loading and maintenance doses and dosing intervals should be based on the patient’s clinical condition, response to therapy and relevant laboratory tests Prothrombinex is supplied as a freeze-dried powder with a 20mL ampoule of water for injection (WFI) and one filter needle. Before reconstitution allow the product to reach room temperature. Using an aseptic technique draw up 20mL of WFI and inject into the vial of Prothrombinex. Dissolve the Prothrombinex by gentle agitation. Do not shake. A clear or slightly opalescent solution is obtained within 10 minutes. If clots or a gel form return the product to the Blood Bank. Infuse slowly intravenously approximately 3mL/minute. |
|
|
MH57 [Jul09]
Patient over-warfarinised and is for surgery. Prothrombinex 50U/kg may NOT reverse an INR of 5.5 because it contains: A. Citrate B. Heparin C. Anti-thrombin III D. Not enough Factor VII E. Not enough Factor X |
ANSWER D
Warfarin inhibits the synthesis of 2, 7, 9, 10 Prothrombin X contains 2, 9 ,10, it has little to no Factor 7 |
|
|
41.(NEW) Patient with Hemophilia A with known high titres of inhibitors to factor 8. What would you give to prevent bleeding in the patient for ot
a. FVIIa b. High dose FVIII concentrate c. FFP d. Cryo e. Platelets |
ANSWER A
Hemophilia A (HA) is an X-linked congenital bleeding disorder resulting from a deficiency of factor VIII (fVIII). Treatment is to replace F8. However, patients may develop inhibitory IgG antibodies to F8. Inhibitor formation occurs in 36% of severe haemophila A patients Treatment of patients with an inhibitor -when first detected in low titres, F8 can still be used -on 5BU/ml of inhibitor detected, F* replacement becomes useless -recombinant activated factor 7 (rfVIIa, novoseven) is used, facilitates haemostasis by activating facotr X directly on platelet surface -halflife is 2.3 hours |
|
|
MH59 [Mar10]-[Aug10]
70 year old post TKJR. On sub-cut heparin. Develops clinical DVT and platelets 40 (sounds like HITS type-II). Management A. Enoxaparin B. Fondoparinux C. Heparin by infusion D. Lepirudin E. Warfarin |
ANSWER D
Management of HIT: * First task is to discontinue unfractionated heparin from ALL sources (including heparin-coated lines, etc). LMWH can also cause HIT, therefore not suitable as a replacement. Fondaparinux is an indirect Factor-Xa inhibitor (synthetic pentasaccharide), and there are some reports of it being used in HIT successfully. Warfarin (Vit K antagonist) is contraindicated in acute HIT (or if suspected HIT), as it can cause skin necrosis or venous limb gangrene. * Current recommendations are to treat with DTI's (lepirudin, argatroban, bivalirudin) or danaparoid. Although danaparoid is a LMW heparinoid, there is an extremely low cross-reactivity rate with HIT antibodies, and this is rarely clinically significant. o As danaparoid is not an option, the best answer is therefore a direct thrombin inhibitor (DTI), and lepirudin is the only one listed, so answer is D. |
|
|
MH61 [Mar2011] A 35yr old African-American with sickle cell and fractured ankle for ORIF. Hb 90, Hct 0.3.
A. T/F 2 units packed cells (?pre-op) B. Let him cool passively to low/normal temperature C. Spinal is safe D. Avoid thiopentone E. Tourniquet is absolutely contra-indicated |
ANSWER C
|
|
|
MH60
ANZCA version [Apr08] Q117 Haemophilia A is commonly associated with: A. a haemarthrosis in a female infant B. a haemarthrosis in a male infant C. low levels of Factor IX D. normal prothrombin time (PT) and prolonged activated partial thromboplastin time (APTT) E. prolonged prothrombin time (PT) and prolonged activated partial thromboplastin time (APTT) |
ANSWER D
D - True (as extrinsic pathway is unaffected, PT is normal) |
|
|
MH59 [Mar10]-[Aug10]
70 year old post TKJR. On sub-cut heparin. Develops clinical DVT and platelets 40 (sounds like HITS type-II). Management A. Enoxaparin B. Fondoparinux C. Heparin by infusion D. Lepirudin E. Warfarin |
ANSWER D
HITS Management of HIT: * First task is to discontinue unfractionated heparin from ALL sources (including heparin-coated lines, etc). LMWH can also cause HIT, therefore not suitable as a replacement. Fondaparinux is an indirect Factor-Xa inhibitor (synthetic pentasaccharide), and there are some reports of it being used in HIT successfully. Warfarin (Vit K antagonist) is contraindicated in acute HIT (or if suspected HIT), as it can cause skin necrosis or venous limb gangrene. * Current recommendations are to treat with DTI's (lepirudin, argatroban, bivalirudin) or danaparoid. Although danaparoid is a LMW heparinoid, there is an extremely low cross-reactivity rate with HIT antibodies, and this is rarely clinically significant. |
|
|
PZ68a [Apr97] [Jul97] [Apr98]
Heparin-induced thrombocytopenia (HITS): A. Rarely see platelets less than 100 B. Thrombocytopaenia will continue when heparin is ceased C. Onset within 2 to 3 days of starting heparin D. Is associated with intravascular thrombosis E. Non-specific antibodies F. Antibodies remain indefinitely |
A - FALSE the clinically important form of the disease frequently does result in much lower counts
B - FALSE - will not continue indefinitely, and once the heparin concentration drops the process of platelet activation stops allowing platelet counts to recover. C - FALSE - it implies if the counts are ok on day 3 you're out of the woods which is definitely untrue and once again the clinically relevant thing the examiner wants you to know True - 90% of HIT is type I which has an onset of 2-3 days D - True/False - 10% of HIT is type II which has 30-80% thrombosis. Type I not associated with thrombosis. E - FALSE o HIT Type II + Immunogenic heparin-PF4 complexes which cause an immunologic response + Antibodies are generated resulting in a complex forming between antibodies, heparin, and PF4 (mediated through the FcyIIa portion of the platelet). This complex leads to further platelet activation resulting in formation of microparticles and thrombin generation. + Antibodies also recognize PF4 bound to heparin on the endothelial surface and this surface becomes activated leading to another route of thrombin production. F - FALSE - The antibodies that are formed may persist for weeks to months following heparin administration |
|
|
PZ68b ANZCA version [2002-Aug] Q93, [2003-Apr] Q58, [2004-Apr] Q49, [2004-Aug] Q80, [Mar06] Q64, [Jul06] Q12
Immunologically mediated heparin-induced thrombocytopaenia is characterised by A. onset within a few days of first starting heparin B. intravascular thromboses C. platelet count rarely reduced below 100x10^9/L D. continuation of thrombocytopaenia after cessation of heparin E. presence of non-specific (heparin-independent) platelet antibodies |
ANSWER B
A. onset within a few days of first starting heparin - FALSE : "A second form of HIT, HIT type II or immune-mediated HIT, demands more attention. In patients receiving heparin for more than 5 days, antibodies to the heparin-platelet factor 4 complex can form, which are capable of binding to platelet Fc receptors and inducing platelet activation and aggregation." (Stoelting Ch17) B. intravascular thromboses - TRUE : "In vivo, this leads to both an increased clearance of platelets with resultant thrombocytopenia and venous and/or arterial thrombus formation, with the potential for severe organ damage (loss of limbs, stroke, myocardial infarction) as well as unusual sites of thrombosis (adrenal, portal vein, skin)." C. platelet count rarely reduced below 100x10^9/L - FALSE D. continuation of thrombocytopaenia after cessation of heparin - FALSE : will not continue indefinitely, and once the heparin concentration drops the process of platelet activation stops allowing platelet counts to recover. E. presence of non-specific (heparin-independent) platelet antibodies - FALSE |
|
|
PZ68c ANZCA version [2003-Aug] Q128, [2005-Apr] Q90, [2005-Sep] Q67
Heparin Induced Thrombocytopenia (HITS) A. is associated with antibodies to complexes of Antithrombin 3 (ATIII) and heparin B. is associated with a more rapid drop in platelet count if the patient has been exposed to heparin within the last three months C. is not associated with the use of low molecular weight heparins D. results in the maintenance of heparin-dependent antibody levels indefinitely after their development E. results in thrombotic complications in most patients |
ANSWER B
A. FALSE : heparin binds to platelet factor 4 which together forms a neoantigen which causes a humoral immune response. ATIII has nothing to do with it. B. TRUE : 'Rapid onset' HIT is assoc with a drop in platelet count within minutes to hours of heparin exposure and affected patients will usually have been exposed to heparin within the last 3 months. C. FALSE : Low molecular weight heparins can cause HIT, and when used in its treatment there is a significant risk of recurrent or progressive thrombocytopenia with or without thrombosis D. FALSE : The antibodies to heparin-platelet factor 4 become undetectable within 50 to 85 days on stopping heparin exposure. E. FALSE : Less than 10% of those who develop an antibody to the heparin-PF4 complex will exhibit a thrombotic event. However the risk varies considerably with the clinical situation and can reach 40% or more in the postop setting when high circulating levels of both activated platelets and thrombin are present, for example, following orthopedic surgery |
|
|
MH32 ANZCA version [2004-Apr] Q21, [2004-Aug] Q46, [2005-Sep] Q16, [Mar06] Q39
Iron deficiency is characterised by A. high serum ferritin level and low serum iron B. high serum ferritin level and absent bone marrow iron C. increased serum ferritin level and normal serum iron D. low serum ferritin level and low serum iron E. low serum iron level and lowered total iron binding capacity |
ANSWER D
Iron Deficency Anaemia -hypochromic and micocytic Decreased -plasma iron -ferritin -marrow depleted of iron stores Increased -transferrin |
|
|
MH32 Black Bank version [Aug96] [Apr97] [Jul97]
Iron deficiency anaemia: A. Increased serum ferritin, decreased serum Fe B. Decreased serum ferritin, increased serum Fe C. Decreased serum ferritin, decreased serum Fe D. Absence of Fe in bone marrow E. Decreased TIBC |
ANSWER C and D
Iron Deficency Anaemia -hypochromic and micocytic Decreased -plasma iron -ferritin -marrow depleted of iron stores Increased -transferrin |
|
|
MH36a ANZCA version [2002-Mar] Q108, [2002-Aug] Q135 (Similar question reported in [Jul97] [Apr98] [Jul98])
Conditions which are associated with a high risk of thromboembolism post-operatively include 1. protein C deficiency 2. protein S deficiency 3. antithrombin III deficiency 4. lupus anticoagulant |
ANSWER ALL
There is an increased risk of thrombosis with: * Protein C deficiency * Protein S deficiency * Antithrombin III deficiency * Lupus anticoagulant and/or anticardiolipin antibody * Factor V Leiden mutation * Thrombocytosis * Polycythemia |
|
|
MH36b ANZCA version [2001-Apr] Q142, [2001-Aug] Q98
Conditions which predispose patients to deep venous thrombosis include 1. antiphospholipid syndrome (lupus anticoagulant) 2. Protein C deficiency 3. Factor V Leiden mutation 4. Protein S deficiency |
ANSWER ALL
There is an increased risk of thrombosis with: * Protein C deficiency * Protein S deficiency * Antithrombin III deficiency * Lupus anticoagulant and/or anticardiolipin antibody * Factor V Leiden mutation * Thrombocytosis * Polycythemia |
|
|
MH36c ANZCA version [2004-Apr] Q32, [2004-Aug] Q1, [Mar06]
Following major surgery, there is an increased risk of thrombosis, associated with a decrease in A. fibrinogen B. factor VIII coagulant6 C. factor VIII; Ag (von Willebrand related antigen) D. interleukin 6 E. protein C |
ANSWER E
There is an increased risk of thrombosis with: * Protein C deficiency * Protein S deficiency * Antithrombin III deficiency * Lupus anticoagulant and/or anticardiolipin antibody * Factor V Leiden mutation * Thrombocytosis * Polycythemia |
|
|
A thromboelastograph (T.E.G.) result on a 64 yr old female having a portacaval bypass operation
is reported as Coagulation time (r+k) 20min (N 10 - 12 min) Maxiumum Amplitute (MA) 30mm (N 50 - 70 mm) Clot formation rate 35 degrees (N > 50 degrees) These results suggest A. A defect of the intrinsic system only B. 6u FFP would correct the deficit C. The TEG should be repeated with episilon-aminocaproic acid added to the specimen D. presence of increased fibrinolysis E. Possible heparin contamination of the specimen |
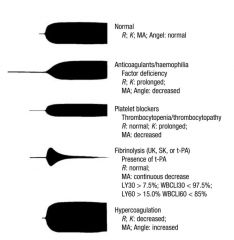
ANSWER E
|
|
|
MH50 ANZCA version [2004-Aug] Q140, [2005-Apr] Q41, [Mar06]
Correct statements regarding desmopressin (DDAVP) include each of the following EXCEPT A. it improves platelet function in patients with liver disease B. it improves platelet function in patients with renal disease C. it is contraindicated in Type 1 von Willebrands disease D. it is given in a dose of 0.3 micrograms.kg’ intravenously E. it is used in the treatment of diabetes insipidus |
ANSWER C
Product information for desmopressin: 1. Dose -DI is 1-4mcg/day (10-40mcg/day nasal). Haemophilia or vWD 0.3-0.4mcg/kg 2. Indications: DI, Haemophilia A, vWD except IIB, platelet disorders 3. May induce platelet aggregation in vWD IIB 4. Congenital or acquired platelet disorders including uraemia, drug induces, CPB induced may respond. No evidence for platelet dysfunction of liver disease and use is not supported. |
|
|
MH52 ANZCA version [2005-Sep] Q130, [Mar06] Q76
The most correct statement describing the effect of the antithrombotic agents on the coagulation cascade is that A. low molecular weight heparin directly inhibits activated factor X B. low molecular weight heparin directly inhibits thrombin C. unfractionated heparin directly inhibits activated factor X D. unfractionated heparin directly inhibits thrombin E. ximelagatran directly inhibits thrombin |
ANSWER E
UFH and LMWH -activate ATIII (increasing it's acitvity 1000 fold) -UFH/ATIII inactivate thrombin and F10a equally -LWMH/ATIII inactivates F10a>thrombin Fondaparinoux -synthetic pentasaccharide -chemically related to heparin -binds and activates ATIII -fonda/ATIII exclusively inactivate F10a -can be used in HITS Ximelagtran -direct thrombin inhibitor -prodrug which is absorbed in SI and converted by the liver to melagatran -removed due to liver toxicity -replaced by dabigatran Dabigatran -oral anticoagulant -direct thrombin inhibitor RELY study -manufacturer sponsered phase III trial -assessor blinded RCT comparing warfarin to dabigatran 110 and 150mg bd in patients with AF -findings -110mg non inferior for stroke and systemic embolization to warfarin -110mg lower risk of bleeding to warfarin -150mg superior for stroke and systemic embolization to warfarin -150mg same risk of bleeding to warfarin RECOVER trial -large, randomized, double blind trial -dabigatran and warfarin in the treatment of acute VTE -non inferior -similar rates of major bleeding -however had more dyspepsia and therefore more drug discontinuation |
|
|
MH53 ANZCA version [2003-Aug] Q125, [2005-Sep] Q91, [Mar06] Q89
In beta-thalassaemia major A. blood transfusion prevents growth retardation in children. B. the red blood cells are hypochromic and macrocytic. C. the average age at death is 25-30 yrs. D. liver failure is the most common cause of death E. iron supplementation improves peri-operative haemoglobin levels. |
ANSWER A
* A. blood transfusion prevents growth retardation in children. - true: "The clinical benefits of hypertransfusion during the first decade of life are dramatic, with reductions in hepatosplenomegaly, partial correction of abnormal skeletal development, and at least short-term improvements in cardiac dilatation and systolic function." (Uptodate accessed 28/7/10) B. the red blood cells are hypochromic and macrocytic. FALSE : hypochromic, microcytic C. the average age at death is 25-30 yrs. D. liver failure is the most common cause of death : FALSE -leading cause of death is iron overload followed by sepsis E. iron supplementation improves peri-operative haemoglobin levels. FALSE Iron supplimentation does not improve haemoglobin levels as the problem is with globin synthesis not haem and the patient usually has too much iron from multiple transfusions. Thalassemia -inherited blood disorder characterised by reduced or absent synthesis of the alpha/beta chains of hemoglobin -severity depends on the degree of impaired globin synthesis Prevalence 1. Mediterranean (mainly b) 2. African (a and b) 3. Asian (mainly a) Diagnosis is conformed by haemoglobin electrophoresis and globin chain analysis Presenation -B-thal major : made at childhood with severe anaemia accompanied by massive ineffective erthropoeisis : splenomegaly, profound microcytosis, elevated elvels of HbF and HbA2. Treatment 1. Long-life blood transfusion to mainly Hct 25-30% which suppresses erythropoisis -results in iron overload and endocrine dysfunction a. glucose intolerance b. thyroid dysfunction c. delayed onset of puberty -also cardiac, liver and pancreas dysfuction 2. Splenectomy Prognosis -poor, death before age 20 -complications due to iron overload |
|
|
MH41 ANZCA version [Jul00] [2001-Aug] Q127, [2002-Mar] Q117, [Jul06] Q46 (now converted to type A version)
Anti-retroviral agents for HIV infection include 1. nucleoside r-transcriptase inhibitors (NRTIs) which are associated with diarrhoea and intravascular volume depletion 2. protease inhibitors which have important effects on the cytochrome P450 system 3. non-nucleoside r-transcriptase inhibitors (N-NRTIs) which may cause elevations in liver function tests 4. protease inhibitors which can lead to problematic hypoglycaemia |
ANSWER 2
|
|
|
MH10b ANZCA version [2005-Apr] Q108, [2005-Sep] Q60, [Jul06] Q18
Sickle cell disease (homozygous haemoglobin SS) is frequently associated with A. cardiomyopathy B. chronic respiratory dysfunction C. nephropathy D. peripheral neuropathy E. all of the above |
ANSWER E
Sickle cell disease -mutation of HbB to for HbS -HbS is biochemically unstable and can precipitate out of solution in the deoxygenated state Heteroxygous -sickle cell trait -30-40%HbS -benign clinical picture but can sickles is stressed -protective again Malaria Homozygous -100%HbS -debilitating -progressive multiorgan damage and early death Clinical presentation (SCD) CVS -cardiomegaly due to anaemia -pulmonary hypertension due to recurrent pulmonary infarcts RESP -dyspnoea, cough, haemoptysis, pleuritic chest pain caused by recurrent pulmonary infaractions -respiratory failure ENT/airway -marrow hyperplasia : front bossing and prominent maxilla -microvascular retinopathy, vitreous haemorrhage and retinal detachment -functional asplenim : hypertrophy of other lymphoid tissues (tonsils and adrenoids) leading to OSA URIN -priaprism -CRI GIT -infarction of spleen : immunie incompetence -gallstones -intrahepatic sicling Skeletal/skin -deformities from marrow hyperplasia -aseptic necrosis and leg ulcers NEURO -TIA -stroke -ACH HAEMO -anaemia -myelosuppresion |
|
|
AB51 ANZCA Version [Jul06] Q103, [Apr07]
A fifty-five-year-old man on antihypertensive medication, including an ACE inhibitor, has a total knee replacement. Red cell transfusion is begun in recovery through a leukocyte reduction filter after brisk bleeding into his drains. A recognised complication of the use of this filter in this situation is A. air embolism B. clotting factor depletion C. haemolysis D. increased risk of postoperative infection E. severe hypotension |
ANSWER E.
severe hypotension - true: "Profound hypotension has been reported in patients taking angiotension-converting-enzyme (ACE) inhibitors and receiving pretransfusion leukocytereduced blood products—platelets in particular (96). Presumably, ACE inhibitors decrease bradykinin degradation thereby prolonging its intravascular half-life." |
|
|
AB52 ANZCA Version [Jul06] Q121
Blood donors most commonly implicated in TRALI (Transfusion Related Acute Lung Injury) are A. diabetics B. donors previously resident in the United Kingdom C. indigenous Australians D. men under 50 years of age E. multiparous females |
ANSWER E
Transfusion-related acute lung injury TRALI is a form of non-cardiogenic pulmonary oedema (ARDS) that occurs following transfusion of blood or blood products. TRALI is most commonly caused by a reaction to leukocyte antibodies present in the plasma component of blood products. These antibodies can activate granulocytes that cause plasma leakage into the lungs, resulting in acute pulmonary edema. According to the Food and Drug Administration, TRALI is a leading cause of transfusion-related deaths in both male and female patients. Multiparous women: The implicated components in TRALI reactions are usually associated with female donors who have had one or more pregnancies or any donor who has been previously transfused. Women are more likely than men to have antibodies to leukocytes due to exposure to fetal antigens during pregnancy. There is currently no screening test for the prevention of TRALI and there is no single intervention that can eliminate the risk of TRALI. So: * Donors with potentially causative antibodies who have been implicated in TRALI reactions are permanently deferred. * Donor leucocyte antibodies are produced mainly as a result of pregnancy. Consequently, transfusing blood from male donors only greatly reduces the risk. In future, all FFP will be sourced from male donors and this should virtually eliminate the risk of TRALI. Presentation TRALI is similar to adult respiratory distress syndrome (ARDS) in its clinical presentation. It presents with: * respiratory distress (dyspnoea, cyanosis), * tachycardia, * fever, and * hypotension. Over several hours the CXR shows 'white-out' with diffuse alveolar and interstitial infiltrates. Symptoms usually arise within 1-6 hours of commencement of transfusion of a plasmacontaining product. Approximately 80% of patients with TRALI improve rapidly (clinically and radiologically) over 48 hours provided there is prompt and vigorous respiratory support. Many patients will require intubation and respiratory support." Diagnostic features * Onset within 1-6 hours of transfusion; * Acute respiratory distress; * Acute bilateral pulmonary edema (noncardiogenic); * Severe hypoxemia; * Hypotension (rarely hypertension); * Fever; * Clinical spectrum: from mild to severe; and * Mortality rate: 6% to 10%. Differential diagnosis Besides TRALI, other transfusion-related complications need to be evaluated in the differential diagnosis, including * circulatory overload (TRACO), * bacterial contamination, and * acute hemolytic transfusion reactions. At variance with circulatory overload that is associated with increased central venous pressure, TRALI is usually not associated with cardiac dysfunction (noncardiogenic pulmonary edema). |
|
|
PZ80c ANZCA version [2001-Aug] Q145
Compared to unfractionated heparin, low molecular weight heparins (LMWH) 1. have a higher bioavailability 2. are more easily reversed with protamine 3. have less effect on platelet function 4. have a shorter biological half life |
1. True
2. False 3. True 4. False |
|
|
PZ80d ANZCA version [2002-Aug] Q44, [2003-Apr] Q27, [Jul06] Q32
The plasma half-life of low molecular weight heparin is A. increased in conditions with raised plasma proteins B. 2 to 4 times that of unfractionated heparin C. much less predictable than unfractionated heparin D. dependent upon a saturatable mechanism for clearance E. longer than unfractionated heparin because of a higher affinity for plasma protein |
ANSWER B
LMW Heparin * Higher bioavailability * Bioavailability is greater. Protein binding is much less compared to unfractionated heparin, giving superior bioavailability at lower doses. * Not fully reversible with protamine unlike unfractionated heparin * Less HITS and less platelet function affect * Longer T 1/2 and more predictable kinetics * clearance by first order kinetics, through the renal route |
|
|
PZ80e ANZCA version [2003-Aug] Q110
Low molecular weight heparins A. are cleared principally by the liver B. are ineffective if only administered post-operatively C. cross the placenta D. have a dose-dependent half-life E. have a longer half-life than standard (unfractionated) heparin |
ANSWER E
Low molecular weight heparins * A. are cleared principally by the liver - false: "Metabolic breakdown of Clexane is slight and takes place mainly in the liver (desulfation and depolymerisation)." (MIMs online) * B. are ineffective if only administered post-operatively - false * C. cross the placenta - false: "The anti-Xa activity generated by Clexane does not cross the placental barrier during the second trimester of pregnancy." (MIMs online) Also "In contrast to warfarin, heparin does not cross the placenta and has not been associated with fetal malformations; therefore it is the drug of choice for anticoagulation during pregnancy." (Goodman and Gilman) * D. have a dose-dependent half-life * E. have a longer half-life than standard (unfractionated) heparin - true |
|
|
MH54 [Apr07]
von Willebrands type 1: A. Variable increase in bleeding time B. Increased aPTT C. Increased PT D. Haemarthroses E. Delayed bleeding after trauma |
ANSWER A
PT normal APTT normal (25% riased) BT raised |
|
|
MH21 ANZCA version [2001-Apr] Q13, [2001-Aug] Q19 (Similar question reported in [1988] [1989] [Mar90] [Sep90] [Jul98])
A patient undergoing suprapubic prostatectomy appears to be bleeding excessively. In an attempt to exclude primary hyperfibrinolysis as a possible cause the most useful test is A clot retraction time B plasma fibrinogen estimation C prothrombin time D thromboelastography E whole blood clotting time |
ANSWER D
Fibrinolysis -breakdown on clot Congential -deficiency in a2 antiplasmin Acquired -liver dysfunction -severe trauma -major surgery -fibrinolytics : streptokinase Diagnosis via TEG APTT and PT are poor Treatment -transaemic acid -aminicaproic acid |
|
|
MH60 ANZCA version [Apr08] Q117
Haemophilia A is commonly associated with: A. a haemarthrosis in a female infant B. a haemarthrosis in a male infant C. low levels of Factor IX D. normal prothrombin time (PT) and prolonged activated partial thromboplastin time (APTT) E. prolonged prothrombin time (PT) and prolonged activated partial thromboplastin time (APTT) |
ANSWER D
A - X-linked, females are usually carriers, B - True for severe haemophilia A, but not exclusively male C - Congenital factor VIII deficiency D - True (as extrinsic pathway is unaffected, PT is normal) E - Normal PT |
|
|
MH56 [Jul09]
Bleeding patient. What is relative contraindication to Prothrombinex? A. History of HITS B. Von Willebrands C. Haemophilia B D. Warfarin overdose ?? or - Overdose vit K (not warfarin) E. Renal failure |
ANSWER A
Prothombinex contains 192 unit of heparin |
|
|
Prothombinex
|
Prothrombinex
-sterile freeze dried powder containing purified human coagulation F2, F9 and F10 (low levels of F5 and F7) -prepared from pooled human plasma -works immediately with 100% bioavailabiity Contents -F9 500 IU -F2 approx 500 IU -F10 approx 500 IU -human plasma proteins (low levels of F5 and F7) -Antithrombin III 25 IU -Heparin 192 IU -NA / PO4 / Citrate / Cl Dose -25-50 IU/kg Contraindications 1. hypersensitivity 2. HITS 3. Active thrombus 4. DIC Risks 1. Thrombus : AMI, CVA, PE 2. DIC 3. viral transmission : CJD 4. skin rash Indications 1. Elevated INR with or without bleeding a. Bleeding where warfarin induced coagulapathy is considered a contributing factor -cease warfarin -5-10mg Vit K IV -Prothrombinex 25-50 IU with FFP -measure INR and bleeding closely b. INR >9 bleeding absent -cease warfarin -1mg Vit K IV -consider PTX and FFP -measure INR closely 2. Invasive procedures -risk of VTE must be considered eg. prosthetic heart valves, recent VTE (<3/12) = these patients should have bridging therapy perioperatively a. Before surgery -Withhold warfarin 4-5 days before surgery -Night of surgery : If INR>2 give 1-5mg Vit K IV -Day of surgery : If INR<1.5 proceed, if INR>1.5 defer surgery, or if urgent give PTX and FFP b. After surgery : start warfarin on day of surgery, resume as previous 3. Congenital or acquired deficiency of Factor 2, 9, 10 4. Haemophilia B |
|
|
MH57 [Jul09]
Patient over-warfarinised and is for surgery. Prothrombinex 50U/kg may NOT reverse an INR of 5.5 because it contains: A. Citrate B. Heparin C. Anti-thrombin III D. Not enough Factor VII E. Not enough Factor X |
ANSWER D
Prothrombinex has 2, 9 and 10 only. Warfarin inhibits 2, 7, 9 and 10. It does contain human plasma proteins < 500mg (incl VII), but VII levels are low and unquantified PTX administration without FFP is recommended only when FFP is unavailable, as PTX factor VII levels are low and unquantified. MIMS Australia. |
|
|
Black bank August 2009 Question 41.
(NEW) Patient with Hemophilia A with known high titres of inhibitors to factor 8. What would you give to prevent bleeding in the patient for ot a. FVIIa b. High dose FVIII concentrate c. FFP d. Cryo e. Platelets |
ANSWER A
The whole reason why novoSeven exists in the first place. A particular therapeutic conundrum is the development of "inhibitor" antibodies against factor VIII due to frequent infusions. These develop as the body recognises the "normal form" factor VIII as foreign, as the body does not have its own "copy". The problem is that in these patients, factor VIII infusions are ineffective. Recently[update] activated factor VII (NovoSeven) has become available as a treatment for haemorrhage in patients with haemophilia and factor inhibitors. |
|
|
TMP-Jul10-002
Male with a haemoglobin of 80%, and a reticulocyte count of 10%. Possible diagnosis: A. Untreated pernicious anaemia B. Aplastic anaemia C. Acute leukaemia D. Anaemia of chronic disease E. Hereditary spherocytosis |
ANSWER E
Hereditary spherocytosis (HS) is a genetically-transmitted (autosomal dominant) form of spherocytosis, an auto-hemolytic anemia characterized by the production of red blood cells that are sphere-shaped rather than donut-shaped, and therefore more prone to haemolysis There is a marked heterogeneity of clinical features, ranging from an asymptomatic condition to fulminant haemolytic anemia. The morphologic hallmark of HS is the microspherocyte, which is caused by loss of membrane surface area, and an abnormal osmotic fragility in vitro. Investigation of HS has afforded important insights into the structure and function of cell membranes and into the role of the spleen in maintaining red blood cell (RBC) integrity. An intrinsic genetic defect causes defects in membrane proteins. The major complications are aplastic or megaloblastic crisis, haemolytic crisis, cholecystitis and cholelithiasis, and severe neonatal haemolysis. Haemolysis in HS results from the interplay of an intact spleen and an intrinsic membrane protein defect that leads to abnormal RBC morphology. HS erythrocytes are caused by membrane protein defects resulting in cytoskeleton instability. |
|
|
TMP-Jul10-047
Young female patient for tonsillectomy. Which causes INCREASED tendency for bleeding? A. Factor V Leiden B. Protein C deficiency C. Haemophilia B (Christmas disease) D. Antithrombin III deficiency E. Lupus anticoagulant |
ANSWER C
Lupus anticoagulant (http://en.wikipedia.org/wiki/Lupus_anticoagulant) is asociated with a tendency to thrombosis. It is a nuisance on coag testing as its leads to an abnormal result which may unnecessarily delay surgery. Factor V Leiden confers resistance to breakdown by protein C & S system. It results in hypercoagulability, and is therefore the wrong answer. Protein C deficiency results in reduced FV & VIII breakdown and hypercoagulability, and is therefore the wrong answer. Haemophilia B, is X-linked but the phenotype in females is variable. It does cause increased bleeding. And is the answer to choose. ATIII deficiency is associated with a hypercoagulable state, and is the wrong answer. Lupus anticoagulant, causes in vitro prolongation of the APTT but in vivo, results in hypercoagulability, and is the wrong answer. |
|
|
H59 [Mar10]-[Aug10]
70 year old post TKJR. On sub-cut heparin. Develops clinical DVT and platelets 40 (sounds like HITS type-II). Management A. Enoxaparin B. Fondoparinux C. Heparin by infusion D. Lepirudin E. Warfarin |
ANSWER D
Management of HIT: * First task is to discontinue unfractionated heparin from ALL sources (including heparin-coated lines, etc). LMWH can also cause HIT, therefore not suitable as a replacement. Fondaparinux is an indirect Factor-Xa inhibitor (synthetic pentasaccharide), and there are some reports of it being used in HIT successfully. Warfarin (Vit K antagonist) is contraindicated in acute HIT (or if suspected HIT), as it can cause skin necrosis or venous limb gangrene. * Current recommendations are to treat with DTI's (lepirudin, argatroban, bivalirudin) or danaparoid. Although danaparoid is a LMW heparinoid, there is an extremely low cross-reactivity rate with HIT antibodies, and this is rarely clinically significant. o As danaparoid is not an option, the best answer is therefore a direct thrombin inhibitor (DTI), and lepirudin is the only one listed, so answer is D. |
|
|
MH09b ANZCA version [2002-Aug] Q41, [2005-Apr] Q83, [2005-Sep] Q85, [Jul07] (Similar question reported in [Mar91] [Mar94] [Aug94] [Aug95] [Apr98])
Anaemia in chronic renal failure is characteristically A. due to haemolysis in the renal vascular bed B. normochromic and microcytic C. due to defective haemoglobin synthesis D. responsive to iron and folate therapy E. associated with increased 2,3-DPG levels in blood cells |
ANSWER E
"The intracellular concentration of 2,3-bisphosphoglycerate is appropriately increased in response to anemia and hyperphosphatemia,9 with a moderate decrease in the affinity of hemoglobin for oxygen." (Williams Hematology) |
|
|
MH61 [Mar2011]
A 35yr old African-American with sickle cell and fractured ankle for ORIF. [Hb] 90g/l, Haematocrit 0.3. A. Transfuse 2 units packed cells (?pre-op) B. Let him cool passively to low/normal temperature C. Spinal is safe D. Avoid thiopentone E. Tourniquet is absolutely contra-indicated |
ANSWER C
|
|
|
MH58 ANZCA version [Jul07]
Heparin induced thrombocytopenia (HITS) A. is associated with antibodies to complexes of anti-thrombin 3 (ATIII) and heparin B. is associated with a more rapid drop in platelet count if the patient has been exposed to heparin within the last 3 months C. is not associated with the use of low molecular weight heparins D. results in the maintenance of heparin-dependent antibodies indefintely after their development E. results in thrombotic complications in most patients |
ANSWER B
A - False * Clinically, there are two types of HIT that can result from heparin administration: HIT type I, a benign non-immune condition in which no heparin-dependent antibodies are present; and HIT type II, a immune-mediated syndrome caused by an antibody to the heparin–platelet factor 4 (PF4) complex B - True Thirty percent of patients present with ‘rapid onset’ HIT type II. They show an abrupt drop in platelet count when heparin is administered. These patients have often received heparin within the last 3 months. C - False * Less binding to PF4 occurs with low molecular weight heparin (LMWH) and pentassacharide, and consequently, these are less antigenic and cause HIT less frequently. D - False * HIT–IgG antibodies are transient and cannot be detected several weeks or months following the episode of HIT type II. HIT–IgG usually are not restimulated despite re-exposure to heparin. In the event that they are re-stimulated, the antibodies develop gradually usually after a 5-day interval E - False (?) * Of the patients who develop HIT–IgG seroconversion, 30–50% will develop thrombocytopenia; and of these, 30–80% will demonstrate isolated thrombotic events, 0.01–0.1% of whom will experience multiple thromboses or ‘white clot syndrome’ o I don't know if this is most...but only refers to HIT II...so overall maybe not that common |
|
|
MH55 ANZCA version [2003-Aug] Q96
Von Willebrand's disease is A. A common acquired defect of platelet function B. Associated with an imbalance of thromboxane A2 and prostacyclin C. not responsive to DDAVP D. Often treated with epsilon aminocaproic acid (Amicar) E. Responsive to FFP |
ANSWER E
A. False. Inherited B. ? Not sure but I don't think that it affects production of either of these. C. Type 2B and 3 are not. These are however rare forms and 80% of VWD is type I which is responsive. D. Emedicine states that this is used for treatment of vWD. Often??? E. Responsive to DDAVP, cryoprecipitate or factor VIII concentrates can be used. FVIII is labile and therefore lower concentrations in FFP. |
|
|
MH47 ANZCA version [2003-Apr] Q149, [2003-Aug] Q1
Prior to elective surgery, an increased bleeding risk is most likely to be identified by A. a platelet count B. a prothrombin time C. a skin bleeding time D. an accurate clinical history E. an examination for bruising |
ANSWER D
The following extract is from Up ToDate: Preoperative assessment of hemostasis (http://www.utdol.com/utd/content/topic.do?file=coagulat/12865&type=A&selectedTitle=3~44) The clinical history — The clinical history remains the cornerstone of the preoperative assessment. However, four reasons have been cited why the history alone may be insufficient, suggesting a role for laboratory testing in some patients [1]: * The forgetful physician — simply put, this is a surgeon who does not take an adequate history. In our current medicolegal climate, this may be a very compelling argument for laboratory screening tests. * The unreliable patient — a common example of the unreliable patient is one who claims to have no prior surgical history but neglects to mention wisdom teeth extractions or tonsillectomy. Both procedures are quite common, often occurring during the childhood years, and hence are often forgotten by the time of adulthood. Both operations are true challenges to hemostasis, since the oral cavity is rich in fibrinolytic substances. Alternatively, patients may recall these procedures but do not consider the bleeding they may have experienced as excessive. Having no basis for comparison, they may inadvertently overlook this complication. Other relevant items often overlooked include circumcision, childbirth and associated surgical procedures (eg, episiotomy), epistaxis, and the extent of menstrual blood losses. * The unprovoked patient — a patient with an inherited bleeding disorder may not have experienced excessive bleeding in the past, especially if there has not been any prior surgery. In such patients the history will be uninformative unless there is a family history of bleeding (eg, mild hemophilia). * The acquired disorder — patients with an acquired bleeding disorder may have no history of excessive surgical bleeding because their hemostatic defect is relatively recent in onset (eg, acquired von Willebrand disease). |
|
|
MH51 ANZCA version [2004-Aug] Q108, [2005-Apr] Q1
Increased bleeding risk is associated with all of the following EXCEPT A. aspirin B. hypothermia C. lupus anticoagulant D. renal failure E. von Willebrand's disease |
ANSWER C
A - True B - True C - False (lupus antiphospholipid → hypercoaguable) therefore an increased clotting risk, not an increased bleeding risk D - True E - True |
|
|
MH46 ANZCA version [2001-Apr] Q129, [2001-Aug] Q123
Desmopressin (DDAVP) 1. is a synthetic analogue of the natural hormone arginine vasopressin 2. has a longer duration of action than antidiuretic hormone 3. causes an increase in Factor VIII, von Willebrand Factor and tissue plasminogen activator 4. is contraindicated in Type IIB von Willebrand’s disease since platelet aggregation may be induced |
ANSWER ALL TRUE
1. True 2. True : DDAVP half-life is 1-3 hours, ADH (and IV vasopressin) 5-20 mins 3. True - ↑ Factor VIII and vWF, tPA effect 4. True - IIb associated with worsening with DDAVP Qu 2 - from my googling,(Katy) |
|
|
MH40 [Jul00] [Apr07] [Jul07]
In a patient who is HIV antibody positive: A. CD4 count commonly 200-400 (units ?cells/ml) B. Cardiomyopathy is a recognised feature C. Toxoplasmosis infection is a common initial presenttion D. Contraindication to epidural blood patch E. Regional anaesthesia should not be used |
ANSWER C
A. FALSE : typically 300-600 in a healthy HIV, <200 is AIDS B. FALSE : toxoplasmosis is one of the AIDS-defining opportunistic infections, but occurs in 8% of AIDS patients C. TRUE D. FALSE : relative contraindication E. FALSE |
|
|
MH39 [Apr99] [Aug99]
Prostacylin: A. Equally efficacious with inhaled NO B. Half-life of 2 to 3 mins C. Platelet (?aggregation/?inhibition) causes it to be contraindicated D. Metabolised in the liver to arachidonic acid E. Improves oxygen saturation & decreases PAP given IV |
ANSWER A
A: T B: T ?Maybe false - Uptodate quotes a half-life of 6 minutes C: F D: F E: F PGI2 is the main arachadonic acid product in vascular endothelium and smooth muscle. It inhibits platelet function, causes vasodilation and releases NO. It has a short half life of 2-3 minutes. For the treatment of pulmonary hypertension, all studies agree that when given via the inhaled route, it is as effective as NO. It has less efficacy when given IV as it causes systemic vasodilation, reducing pulmonary blood flow and resulting in V/Q mismatch with no improvement in SaO2. Given via the inhaled route, it reduces mean pulmonary arterial pressures and improves SaO2. It is metabolised by spontaneous hydrolysis in the plasma, these products then undergoing further metabolism in the liver. It is not re-formed into arachadonic acid. No important effects on coagulation are reported when it is inhaled. |
|
|
MH31 [Aug93]
Vitamin K dependent factors include: A. V B. VII C. VIII D. X |
ANSWER IS B and D
Vit K dependent clotting factors are II, VII, IX and X. |
|
|
MH30 [Aug93] [Mar94]
Prolonged PT can be seen in: A. Vitamin K deficiency B. Liver disease C. Coumarin therapy D. DIC |
ALL TRUE
|
|
|
MH29 [Aug92] [Mar93] [Aug93] [Aug95]
Von Willebrand's disease: A. Autosomal dominant B. Abnormal bleeding time C. Usually abnormal APTT D. Treatment of choice for bleeding is factor VIII |
ANSWER A and B
Von Willebrands disease * Autosomal dominant trait (for Type I and II) and autosomal recessive (Type 3) affecting <3% of population * Deficit or defective amounts of VWF (von Willebrand factor) which is required for adherence of platelets to endothelium * Binding protein for Factor VIII in plasma * Three forms: 1. Type I - Quantitative decrease but normal vWF and Factor VIII. Usually mild clinical symptoms with variable penetrance. 70-80% of VWD is this type. 2. VWD type 2 has a number of different forms but is primarily a qualitative disease of vWF (ie non functional vWF). This is 15-20% of VWD 3. Type 3 - Undetectable vWF; therefore most severe. Types 2 and 3 do NOT respond to DDAVP. 25% of patients with type I have APTT outside reference range, and pts with type II or III who have abnormal vWF and therefore abnormal factor 8 complex will have abnormal aPTT. Replacing factor 8 may help, but vWF is needed as well. Cryoprecipitate or plasma derived concentrates of Factor VIII (doses vary depending on clinical situation) are used. DDAVP may stimulate release of vWF. Note that recombinant preparations of FVIII do not contain vWF and are therefore not effective. * DDAVP (0.3 mcg/kg IV) for mild bleeding * Factor VIII-vWF concentrate recommended for severe or surgical bleeding (40 - 75 IU/kg) - not purified factor VIII * Cryoprecipitate may be used but carries risk of transfusion-transmitted infection * Aim for VIII levels 50 - 70 % |
|
|
MH28 [Mar92] [Aug99]
Thalassaemia (beta-trait) -you would expect: A. Transferrin levels are normal (or ?increased) B. HbF shifts curve to the right C. Hepatosplenomegaly is a common presentation D. Anaemia worse with infection E. Very large amounts of HbF F. Treated with iron |
ANSWER A
A: TRUE * In thalassaemia triat serum ferritin and iron stores are normal...Kumar and Clarke B: FALSE * Left (? due to increased HbA2) C: FALSE * Common in thalassaemia major - represents extra-medullary hamaetopoiesis (along with bone expansion)...Kumar and Clarke D: ? * No ref in Harrison's. I think this is a distractor relating to sickle cell anaemia * Beta-thalassaemia trait (ie heterozygous) may be asymptomatic or mild anaemia worsened in infections or pregnancy. E: FALSE * Hallmark of thalassaemia major F: FALSE * Iron should not be given...Kumar and Clarke No wonder nobody else has tried to answer this! E - true-the film shows very hypochromic, microcytic cells with target cells and nucleated RBCs. increased HbF++ HbA2 variable, HbA absent. |
|
|
MH25 [Mar90] [Sep90] [Mar91] [Mar95]
Anaemia in Chronic Renal Failure:- A. Hypochromic microcytic B. Normochromic microcytic C. Normochromic normocytic D. Hypochromic normocytic E. Hypochromic macrocytic |
ANSWER C
|
|
|
MH24 [1989]
Thalassemia major:- A. Affects HbF more then HbA B. Most die around 25-30 years C. Liver failure is the most common cause of death D. Associated with microcytic normochromic anaemia E. Growth retardation is fixed by transfusion |
ANSWER B
A - False - No HbA, HbF normal B - True - 25-30 years due to cardiac siderosis (Oxford Handbook of Med) * Chronic transfusions with RBCs improves oxygen delivery, suppresses the excessive ineffective erythropoiesis, and prolongs life, but the inevitable side effects, notably iron overload, usually prove fatal by age 30...Harrison's C - False - CCF and chronic hypoxia D - False - microcytic, hypochromic * Hypochromia and microcytosis characterize all forms of beta thalassemia because of the reduced amounts of hemoglobin tetramers...Harrison's E - True - Development normal with adequate transfusion (OxHM) * If these children are transfused, the marrow is “switched off”, and growth and development may be normal...ABC of Clinical Haematology (BMJ Books) * according to Harrison's 14th Ed 651, death in untreated pts is in 2nd decade (i.e. 20-30). Also says that bone deformity can be "largely prevented" by maintaining normal Hb level throughout. |
|
|
MH16 [1986] [Mar95] [Aug95]
Thrombocytopaenia occurs in: A. Hypersplenism B. Massive transfusion C. Disseminated intravascular coagulation D. B12 deficiency |
ALL TRUE
|
|
|
MH15
Which of the following does cause a crisis in a patient with sickle cell anaemia? A. Hypothermia B. Respiratory acidosis C. Metabolic acidosis D. Hypotension E. Hypokalaemia |
ANSWER E
|
|
|
MH14c ANZCA version [2001-Apr] Q62, [2003-Apr] Q146
Patients with sickle-cell trait A. are usually anaemic B. never sickle C. sickle at very low haemoglobin saturations D. sickle with hypercarbia E. sickle with hyperthermia |
ANSWER C
|
|
|
MH13 [Aug91]
Sickle cell disease is: A. Homozygous recessive B. Confined to black Africans C. Autosomal dominant D. NOT contraindication to Bier's block E. Associated with haemoglobin A |
ANSWER A
A. TRUE : autsomal recessive B. FALSE : common in africans C. FALSE : autosomal recessive D. FALSE : IBRA and tourniquests are contraindicated E. FALSE : HbS or HbC |
|
|
MH11
Sickle cell anaemia: A. Occurs in people of central European descent B. Sex-linked inheritance C. Decreased Hb solubility at low pO2 D. OxyHb dissociation curve shifted to left |
ANSWER C
A - FALSE (?) * Central Africa - 25 % carry the allele o SCD originated in West Africa, where it has the highest prevalence * Also occurs in parts of India and the Mediterreanean (Central Europe is typically Germany / Austra / Czech Rep...but this may be just a poorly worded question) B - FALSE * Autosomal C - TRUE * HbS polymerizes at low PaO2 o Also ppt. by cold, acidosis, infection, dehyadration D - FALSE * OxyHb curve shifted right (P50=31mmHg)... |
|
|
MH08 [1988] [1989] [Mar90] [Aug91] [Jul97]
Normochromic normocytic anaemia is associated with: A. Rheumatoid arthritis B. Acute blood loss C. Renal failure D. Hypothyroidism |
ALL TRUE
|
|
|
MH07 [1986] [Apr97]
Which of the following is NOT seen in acute haemoglobinaemia? A. Fever B. Increased bilirubin in the urine C. Increased stercobilinogen in the gut D. Increased urobilinogen E. Reticulocytosis |
ANSWER B
Acute haemoglobinaemia = intravascular haemolysis Features * Fever * Pallor (if anaemic) * Jaundice * Splenomegaly * Urine: * haemoglobinuria * haemosiderinuria * increased urobilinogen * no increased urine bilirubin * Haemoglobinaemia * Methaemalbuminaemia |
|
|
MH06
Which of the following is NOT seen in haemophilia: A. Increased bleeding time B. Increased thrombin time C. Haemarthrosis D. Abnormal thromboplastin generation time |
ANSWER A and B
|
|
|
MH04 [1986] [Mar93] [Aug96]
A patient has Haemoglobin 8G% and a reticulocyte count of 10%. This picture is compatible with: A. Bone marrow suppression B. Leukaemia C. Aplastic anaemia D. Hereditary spherocytosis E. Untreated pernicious anaemia |
ANSWER D
Hereditary spherocytosis is associated with an increased reticulocyte count. None of the others are. |
|
|
MH02 [Sep90] [Mar93] [Aug95] [Apr98] (type A)
A middle-aged woman, slightly jaundiced and with a spleen just palpable, has a Hb of 7G/dl. The blood film report reads as follows: “Erythrocytes show anisocytosis and range in size from microcytes to oval macrocytes, many showing poikilocytosis. They are normochromic. The platelets are diminished in number. There is a leucopenia and a slight neutropenia. The most likely diagnosis is: A. Pernicious anaemia B. Chronic blood loss C. Haemolytic anaemia D. Hepatitis A E. Carcinomatosis |
ANSWER A
|