Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
73 Cards in this Set
- Front
- Back
|
activity of heparin
antidote? |
increase PTT
decrease fibrinogen activates antithrombin III affects intrinsic pathway protamine sulfate |
|
|
activity of warfarin
antidote? |
increase PT
inhibits vitamin K affects extrinsic pathway teratogenic (crosses placenta)! vitamin K |
|
|
activity of enoxaparin
|
a LMWH, inhibits factor Xa
does not have to be monitored QD/BID dosing |
|
|
why should patients being initiated on anticoagulation be started on heparin before warfarin?
|
warfarin inhibits proteins C and S before vitamin K-dependent factors (II, VII, IX, and X) and therefore causes a transient period of paradoxical hypercoagulability before achieving its anticoagulant effects
|
|
|
what is goal INR of anticoagulation?
|
2.0-3.0
(2.5-3.5 if mechanical heart valve) |
|
|
describe the intrinsic and extrinsic pathways of the coagulation cascade
|
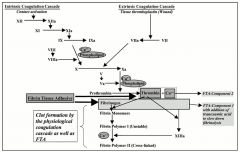
|
|
|
define hemophilia
etiology of this condition? |
deficiency of clotting factor that leads to bleeding diathesis
usually hereditary, but may be acquired through development of antibody to a clotting factor. |
|
|
presentation of hemophilia
|
spontaneous hemorrhage into tissues and joints most commonly
spontaneous intracerebral hemorrhage, renal and retroperitoneal bleeding and GI bleeding may also be seen mild cases may not present until after surgery or trauma |
|
|
lab values to Dx hemophilia
other tests? |
PT: usually normal
aPTT: prolonged (degree of prolongation is gauge of severity) Thrombin, fibrinogen, bleeding time: usually normal mixing study, specific assays for clotting factors (VII, VIII, IX, XI, XII) |
|
|
what levels of clotting factors correspond to hemophilia severity?
|
mild: >5%
moderate: 1-3% severe: <1% |
|
|
treatment for bleeding episodes in hemophilia?
|
immediate transfusion of clotting factors (or cryoprecipitate) to 40% normal
BID Factor VIII (t1/2=12h), QD Factor IX (t1/2=24h) desmopressin in mild cases (fluid restrict to avoid hyponatremia) RBCs may be encessary |
|
|
what is the action of desmopressin (DDAVP) in the treatment of hemophilia?
|
assists in release of extra factor VIII from body
|
|
|
von Willebrand Disease (vWD):
etiology clotting factors affected |
inherited in AD fashion
vWF normally carries factor VIII, but levels are low in vWD and patients are factor VIII deficient the most common inherited bleeding disorder (1% of population affected) |
|
|
presentation of von Willebrand's Disease
|
easy bruising
mucosal bleeding (e.g. epistaxis, oral bleeding) menorrhagia postincisional bleeding NO PETECHIAE, Sx worsen with ASA use |
|
|
coagulation lab results in vWD?
|
PT: normal
aPTT: prolonged ristocetin cofactor assay of patient plasma can measure vWF's capacity to agglutinate platelets |
|
|
treatment of vWD bleeding episodes?
|
DDAVP
menorrhagia can be controlled with OCPs avoid ASAs and other inhibitors of platelet function |
|
|
most common inherited hypercoagulable state?
|
factor V Leiden
a polymorphism in factor V, rendering it resistant to inactivity by activated protein C |
|
|
what are physiologic causes of hypercoagulable states?
|
pregnancy
advanced age |
|
|
what complications are associated with hypercoagulable states?
|
complications are RECURRENT:
DVT, PE, arterial thrombosis, MI, stroke women may have recurrent spontaneous abortions |
|
|
what is the workup for a hypercoagulable patients?
|
lupus antigen/antiphospholipid syndrome
antithrombin III deficiency protein C and S deficiencies activated protein C resistance homocysteine elevation prothrombin G20210A mutation |
|
|
what are the two most common forms of acquired coagulopathy?
|
1. liver disease (most common)
2. DIC |
|
|
what is the characteristic dual nature of DIC?
what is the pathophysiology? |
thrombosis AND hemorrhage
caused by deposition of fibrin in small blood vessels, leading to thrombosis and end-organ damage; depletion of clotting factors leads to a bleeding diathesis may be associated with almost any severe illness |
|
|
acute v. chronic DIC: presentation
|
acute: generalized bleeding out of venipuncture sites, into organs; ecchymoses and petechiae
chronic: bruising and mucosal bleeding, thrombophlebitis, transient neurologic syndromes |
|
|
acute v. chronic DIC: lab values
|
acute: PT, aPTT, thrombin time, and D-dimer are ELEVATED
platelets and clotting factors are DECREASED chronic: PT, aPTT, thrombin time, and D-dimer are ELEVATED platelets and clotting factors are NORMAL |
|
|
thrombotic thrombocytopenis purpura (TTP): related diseases
|
hemolytic-uremic syndrome (HUS)
hemolysis, elevated liver enzymes, and low platelet (HELLP) syndrome |
|
|
TTP: pathogenesis
|
platelet microthrombi (uncertain origina) block of small blood vessels leading to end-organ ischemia and dysfunction; RBCs lysed by contact with microthrombi (microangiopathic hemolytic anemia)
|
|
|
TTP: history
|
clinical syndrome; 5 S/Sx
1. low platelets 2. microangiopathic hemolytic anemia 3. neurologic changes (delirium, seizure, stroke) 4. impaired renal function 5. fever |
|
|
TTP: diagnosis
|
assess for the 5 clinical signs (it is rare that all are present)
assess for schistocytes/nucleated RBCs on peripheral blood smear labs for hemolytic anemia (high indirect bili, LDH, AST; low haptoglobin) coagulation labs are normal |
|
|
severe elevation of Cr: more common in TTP or HUS?
|
HUS, which is characterized by renal FAILURE (v. impairment in TTP)
|
|
|
how to distinguish DIC v. TTP?
|
coagulation studies: PT and aPTT prolonged in DIC, normal in TTP
|
|
|
TTP: treatment
|
corticosteroids, plasma replacement, plasmapheresis
DO NOT GIVE PLATELETS |
|
|
idiopathic (immune-mediated) thrombocytopenic purpura: basic characteristics
|
characterized by presence of anti-platelet IgG antibodies
causes decreased platelet count; bone marrow responds by increasing platelet production with increased megakaryocytes seen in the BM THIS IS THE MOST COMMON IMMUNOLOGIC DISORDER IN WOMEN OF CHILDBEARING AGE |
|
|
ITP: typical patient history and associated diseases
|
patients often feel well; no systemic Sx
minor mucocutaneous bleeding, easy bruising associated with lymphoma, leukemia, SLE, HIV, HCV |
|
|
ITP: presentation
|
may be acute or chronic
acute: abrupt onset of hemorrhage following viral illness (commonly affecting kids 2-6 years) chronic: insidious onset not related to infection; affects primarily women ages 20-40 |
|
|
ITP: diagnosis
|
Dx of exclusion; rule out other causes of thrombocytopenia first
|
|
|
ITP: treatment
|
Tx is reserved for patients with spontaneous bleeding:
corticosteroids, high-dose IVIG; splenectomy if refractory to these |
|
|
anemia: definition
MCV values to classify anemia |
low Hct and Hb
microcytic: <80 fL normocytic: 80-100 fL macrocytic: >100 fL |
|
|
iron deficiency anemia:
give MCV. who is most commonly affected? |
<80 (microcytic)
toddlers, adolescent girls, women of childbearing age, anyone with chronic GI blood loss |
|
|
iron deficiency anemia: Sx
|
fatigue
weakness brittle nails pica patients may be asymptomatic if the anemia develops slowly |
|
|
iron deficiency anemia: physical exam findings
|
glossitis
angular cheilitis koilonychia (spoon nails) |
|
|
iron deficiency anemia: Dx
|
BM biopsy is gold standard but rarely done
iron studies (iron, TIBC/transferrin, ferritin, serum transferrin receptor) blood smear showing hypochromic, microcytic RBCs |
|
|
iron deficiency anemia versus anemia of chronic disease: labs
|
iron deficiency anemia:
iron -- L TIBC/transferrin -- H ferritin -- L serum transferrin receptor -- H anemia of chronic disease: iron -- L TIBC/transferrin -- L ferritin -- H serum transferrin receptor -- normal if both conditions present, looks more like iron deficiency: iron -- L TIBC/transferrin -- normal or H ferritin -- normal or L serum transferrin receptor -- normal or H |
|
|
iron deficiency anemia: Tx
|
replacement iron for 4-6 months
oral iron may cause GI problems; antacids may interfere with iron absorption |
|
|
why do B12 and folate deficiency lead to megaloblastic anemia?
|
deficiency interferes with DNA synthesis, leading to a delay in blood cell maturation and increase in blood cell size
|
|
|
what conditions lead to deficiency of B12 and folate?
|
B12: usually pernicious anemia (destruction of parietal cells and thus lack of intrinsic factor
folate: dietary insufficiency, malabsorption, alcoholism, certain drugs drugs that interfere with DNA synthesis (e.g. some chemotherapeutics) may lead to megaloblastic anemia |
|
|
presentation of B12 (cobalamin) or folate deficiency
|
fatigue
pallor diarrhea loss of appetite headaches tingling/numbness of hands and feet B12 (cobalamin) deficiency may lead to a demyelinating disorder with motor, sensory, autonomic and/or neuropsychiatric dysfunction (subacute combined degeneration of the cord) |
|
|
B12 or folate deficiency: Dx
|
peripheral smear shows elevated MCV; hypersegmented granulocytes might be present
|
|
|
B12 versus folate deficiencies: levels of methylmalonic acid (MMA) and homocysteine
|
B12 deficiency: elevated MMA, elevated homocysteine
folate deficiency: normal MMA, elevated homocysteine |
|
|
hemolytic anemia: typical MCV
|
80-100 fL (it is a normocytic anemia!)
|
|
|
hemolytic anemia: etiologies
|
G6PD deficiency (X-linked recessive transmission)
paroxysmal nocturnal hemoglobinuria hereditary spherocytosis autoimmune RBC destruction (after EBV or mycoplasma infection, CLL, rheumatoid arthritis, medications) sickle cell disease microangiopathic hemolytic anemia (TTP, HUS, DIC) mechanical hemolysis malaria, hypersplenism |
|
|
hemolytic anemia: presentation
|
pallor
fatigue tachycardia tachypnea patients typically jaundiced, with low haptoglobin and elevated indirect bilirubin, LDH dark urine of hemoglobinuria elevated reticulocyte count |
|
|
hemolytic anemia: Tx
|
corticosteroids, iron supplementation
steroids address immunologic causes, iron replaces urinary losses; splenectomy or transfusion in refractory/severe cases |
|
|
problem in aplastic anemia and underlying cause
|
destruction of bone marrow cells means failure of blood cell production
|
|
|
etiology of aplastic anemia
|
multiple:
hereditary (Fanconi) autoimmune viral (HIV, Parvovirus B19) toxins radiation |
|
|
history/exam in aplastic anemia
|
pallor
weakness tendency to infection (most dangerous feature!) petechiae bruising bleeding |
|
|
Dx of aplastic anemia
|
clinical presentation and CBC
verification with BM biopsy |
|
|
physical exam in Fanconi anemia
|
café au lait spots
short stature radial/thumb hypoplasia/aplasia |
|
|
aplastic anemia: Tx
|
blood transfusion
stem cell transplant cyclosporin A + anti-thymocyte globulin (immunosuppress) |
|
|
sickle cell disease (SCD): transmission and underlying defect
|
AR inheritance; mutation in adult hemoglobin (glu-val missense in Beta chain of globin)
|
|
|
characteristics of RBCs in SCD
|
decreased RBC survival
tendency of sickled cells to vaso-occlusion |
|
|
early complications of SCD
|
asymptomatic in first two years of life
later hemolysis results in anemia, jaundice, cholelithiasis, high cardiac output (--> murmur and cardiomegaly), and delayed growth |
|
|
why does spleen auto-infarct in SCD?
|
chronic vaso-occlusion by sickled cells leads to ischemic organ damage and infarction
|
|
|
common triggers of vaso-occlusive crises in SCD
|
cold temperatures
dehydration infection |
|
|
what is common trigger of aplastic crisis in SCD?
|
parvovirus B19 infection
|
|
|
Dx of SCD
|
blood smear shows sickle cells and target cells
gold standard is quantitative Hb electrophoresis |
|
|
mechanism of hydroxyurea in SCD
|
stimulates Hb F production
hydroxyurea is teratogenic! |
|
|
Tx for SCD and complications
|
hydroxyurea
chronic transfusion therapy cholecystectomy for cholelithiasis VOCs: pain management, O2, IV fluid rehydration, abx |
|
|
Dx of thalassemias
|
Hb electrophoresis
|
|
|
Why are chelators (e.g. deferoxamine) given to transfusion dependent beta-thalassemia major patients?
|
to prevent iron overload
|
|
|
"hyperviscosity syndrome" features
|
easy bleeding/bruising
blurred vision neurologic abnormalities plethora pruritus hepatosplenomegaly CHF |
|
|
causes of primary erythrocytosis?
|
hypoxia (lung disease, smoking, altitude)
polycythemia vera (clonal proliferation of pluripotent stem cell) |
|
|
Treatment for polycythemia vera?
|
phlebotomy for symptoms
ASA (for thromboprophylaxis) cytoreductive drugs such as hydroxyurea or IFN |
|
|
Tx for transfusion reactions
|
First, stop the transfusion. Then:
nonhemolytic febrile: Tylenol minor allergic: antihistamines (then epi, then steroids) hemolytic: volume replacement, diuretics and pressors |