Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
240 Cards in this Set
- Front
- Back

What kind of RBC is this? What pathology is associated with this? |

Acanthocyte (Spur Cell)
- Liver disease - Abetalipoproteinemia (state of cholesterol dysregulation |
|
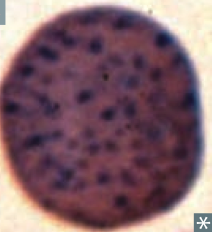
What kind of RBC is this? What pathology is associated with this?
|
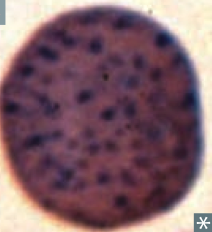
Basophilic Stippling
- Anemia of chronic disease - Alcohol abuse - Lead poisoning - Thalassemias "BASically, ACiD alcohol is LeThal" |
|
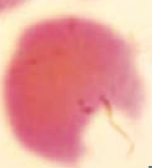
What kind of RBC is this? What pathology is associated with this?
|
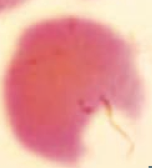
Bite Cell
- 6GPD Deficiency |
|
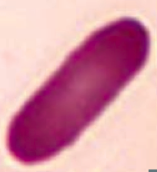
What kind of RBC is this? What pathology is associated with this?
|
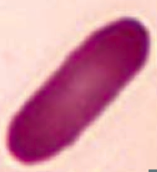
Elliptocyte
- Hereditary elliptocytosis |
|
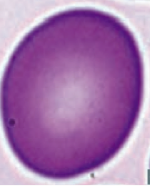
What kind of RBC is this? What pathology is associated with this?
|
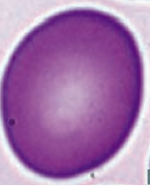
Macro-Ovalocyte
- Megaloblastic anemia (also associated with hypersegmented PMNs) - Marrow failure |
|
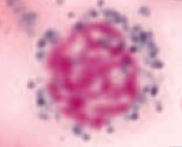
What kind of RBC is this? What pathology is associated with this?
|
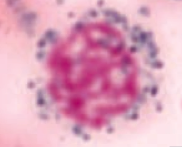
Ringed Sideroblast
- Sideroblastic anemia - Excess iron in mitochondria = pathologic |
|

What kind of RBC is this? What pathology is associated with this?
|

Schistocyte / Helmet Cell
- DIC, TTP / HUS - Traumatic hemolysis (ie, mechanical heart valve prosthesis) |
|

What kind of RBC is this? What pathology is associated with this?
|

Sickle Cell
- Sickle cell anemia |
|
What kind of RBC is this? What pathology is associated with this?
|
Spherocyte
- Hereditary spherocytosis - Auto-immune hemolysis |
|
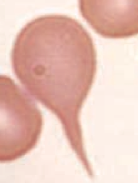
What kind of RBC is this? What pathology is associated with this?
|
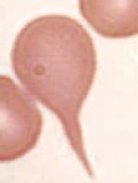
Teardrop cell
- Bone marrow infiltration (eg, myelofibrosis) - RBCs shed a tear because it's forced out of its home in the BM |
|

What kind of RBC is this? What pathology is associated with this?
|

Target Cell
- HbC disease - Asplenia - Liver disease - Thalassemia "HALT" said the hunter to his target |
|
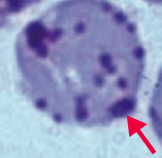
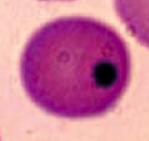
What kind of RBC pathology is this? What causes its formation? What pathology is associated with this?
|
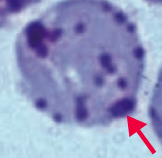
Heinz Bodies
- Oxidation of hemoglobin sulfhydryl groups → denatured hemoglobin precipitation and phagocytic damage to RBC membrane → bite cells - Visualized with special stains such as crystal violet * Seen in G6PD deficiency * Heinz body-like inclusions seen in α-thalassemia |
|
What kind of RBC pathology is this? What causes its formation? What pathology is associated with this?
|
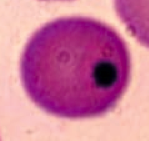
Howell-Jolly Bodies
- Basophilic nuclear remnants found in RBCs - Normally removed from RBCs by splenic macrophages * Seen in patients with functional hyposplenia or asplenia |
|
|
How can you categorize the types of anemias?
|
- Microcytic (MCV <80 fL)
- Normocytic (MCV 80-100 fL) - Macrocytic (MCV >80 fL) |
|
|
What are the types of microcytic anemias? Definition?
|
MCV <80 fL
- Iron deficiency (late) - Anemia of chronic disease (may initially present as a normocytic anemia) - Thalassemias - Lead poisoning - Sideroblastic anemia |
|
|
What are the types of normocytic anemias? Definition?
|
MCV 80-100 fL
Non-hemolytic (reticulocyte count normal or ↓) Hemolytic (reticulocyte count ↑) - Intrinsic - Extrinsic |
|
|
What are the types of non-hemolytic, normocytic anemias? Definition?
|
MCV 80-100 fL and reticulocyte count normal or ↓
- Anemia of chronic disease (may progress to microcytic anemia) - Aplastic anemia - Chronic kidney disease - Iron deficiency (early, may progress to microcytic) |
|
|
What are the types of intrinsic hemolytic, normocytic anemias? Definition?
|
MCV 80-100 fL with increased reticulocyte count
- RBC membrane defect (hereditary spherocytosis) - RBC enzyme deficiency (G6PD or pyruvate kinase) - HbC defect - Paroxysmal nocturnal hemoglobinuria - Sickle cell anemia |
|
|
What are the types of extrinsic hemolytic, normocytic anemias? Definition?
|
MCV 80-100 fL with increased reticulocyte count
- Auto-immune - Microangiopathic - Macroangiopathic - Infections |
|
|
What are the types of macrocytic anemias? Definition?
|
MCV >100 fL
- Megaloblastic - Non-megaloblastic |
|
|
What are the types of macrocytic, megaloblastic anemias? Definition?
|
MCV >100 fL
- Folate deficiency - B12 deficiency - Orotic aciduria |
|
|
What are the types of macrocytic, non-megaloblastic anemias? Definition?
|
MCV >100 fL
- Liver disease - Alcoholism - Reticulocytosis |
|
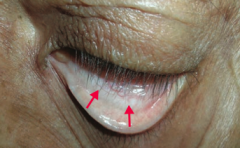
What is the most likely cause of this patient's conjunctival pallor?
|
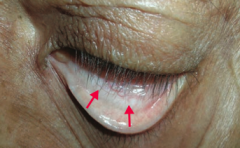
Anemia (possibly due to iron deficiency)
|
|
|
What causes iron deficiency anemia?
|
Decreased iron due to:
- Chronic bleeding (eg, GI loss or menorrhagia) - Malnutrition / absorption disorder - ↑ Demand (eg, pregnancy) |
|
|
What are the implications of an iron deficiency?
|
Decreased completion of final step in heme synthesis
|
|
|
What lab findings are associated with iron deficiency anemia?
|
- MCV < 80 fL (microcytic)
- ↓ Iron - ↑ TIBC - ↓ Ferritin - Hypochromia |
|
|
What other unrelated symptoms should you look for in a patient with iron deficiency anemia to check for another syndrome?
|
Plummer-Vinson Syndrome:
- Also esophageal webs and atrophic glossitis |
|
|
What is the term for the triad of iron deficiency anemia, esophageal webs, and atrophic glossitis?
|
Plummer-Vinson Syndrome
|
|
|
What are the symptoms in Plummer-Vinson Syndrome?
|
- Iron deficiency anemia
- Esophageal webs - Atrophic glossitis |
|
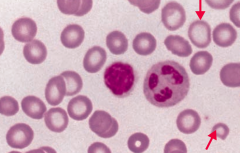
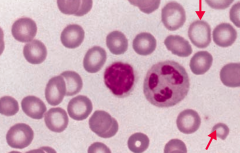
What does this blood smear tell you?
|
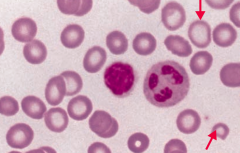
Iron Deficiency Anemia
- Microcytosis - Hypochromia (central pallor) |
|
|
What is the defect in α-thalassemia?
|
α-globin gene deletions → ↓ α-globin synthesis
|
|
|
What kind of deletions can lead to α-thalassemia?
|
- Cis deletion prevalent in Asian populations
- Trans deletion prevalent in African populations |
|
|
How many copies of the α-globin gene do you have? Implications?
|
4 alleles:
- 1-2 allele deletion → no significant anemia - 3 allele deletion → HbH disease, very little α-globin, excess β-globin forms β4 (HbH) - 4 allele deletion → no α-globin, excess γ-globin forms γ4 (Hb Barts); incompatible with life (causes hydrops fetalis) |
|
|
What causes hydrops fetalis?
|
4 α-globin allele deletion:
- No α-globin - Excess γ-globin forms γ4 (Hb Barts) - Incompatible with life |
|
|
What causes HbH disease?
|
3 α-globin allele deletion:
- HbH disease, very little α-globin - Excess β-globin forms β4 (HbH) |
|
|
What form of α-thalassemia is more common in Asians?
|
Cis deletion of α-globin alleles
|
|
|
What form of α-thalassemia is more common in Africans?
|
Trans deletion of α-globin alleles
|
|
|
Who is more likely to have β-thalassemia?
|
Prevalent in Mediterranean poulations
|
|
|
What causes β-thalassemia?
|
Point mutations in splice sites and promoter sequences → ↓ β-globin synthesis
|
|
|
What are the types of β-thalassemia?
|
- β-thalassemia minor (heterozygote)
- β-thalassemia major (homozygote) - HbS / β-thalassemia heterozygote |
|
|
What is the most severe form of β-thalassemia?
|
β-thalassemia major (homozygote)
- β chain is absent → severe anemia |
|
|
What are the consequences of having absent β chain (β-thalassemia major)?
|
- Severe anemia → requires blood transfusion
- Marrow expansion → skeletal deformities → "crew cut" on skull x-ray - Chipmunk facies - Extramedullary hematopoiesis → hepatosplenomegaly - ↑ Risk of parvovirus B19-induced aplastic crisis |
|
|
What kind of infection are patients with β-thalassemia major at risk for? Complications?
|
Parvovirus B19 → can induce an aplastic crisis
|
|
|
What kind of hemoglobin is more common in patients with β-thalassemia major?
|
HbF (α2γ2) - protective in the infant and disease only becomes symptomatic after 6 months
|
|
|
What is the medium severity form of β-thalassemia?
|
HbS / β-thalassemia heterozygote
- Mild to moderate sickle cell disease depending on the amount of β-globin production |
|
|
What is the least severe form of β-thalassemia?
|
β-thalassemia Minor (heterozygote)
- β chain is underproduced, usually asymptomatic |
|
|
How do yo confirm diagnosis of β-thalassemia minor (heterozygote)?
|
Confirm diagnosis by ↑ HbA2 (>3.5% on electrophoresis)
|
|
|
What happens to RBCs in patients with β-thalassemia major?
|
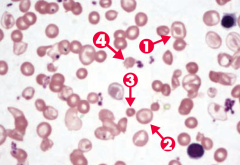
Note anisocytosis, poikilocytosis, target cells (arrows 1 and 2), microcytosis (arrow 3), and schistocytes (arrow 4)
|
|
|
How does lead poisoning affect the blood?
|
- Lead inhibits ferrochelatase and ALA dehydratase → ↓ heme synthesis and ↑ RBC protoporphyrin
- Also inhibits rRNA degradation, causing RBCs to retain aggregates of rRNA (basophilic stippling) |
|
|
Who is at risk for lead poisoning?
|
High risk in old houses with chipped paint
|
|
|
How does lead poisoning affect other organs besides the blood?
|
LEAD:
- Lead Lines on gingivae and on metaphyses of long bones on x-ray - Encephalopathy and Erythrocyte basophilic stippling - Abdominal colic and sideroblastic anemia - Drops: wrist and foot drop |
|
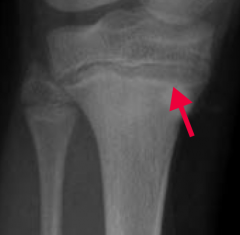
What does the arrow point at? Sign of?
|
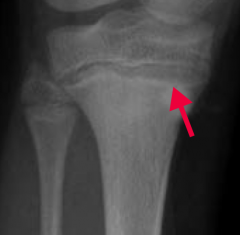
- Lead Lines on metaphyses of long bones on x-ray
- Sign of Lead Poisoning |
|
|
How does lead affect the gums?
|
Lead Lines on gingivae = Burton lines
- Sign of lead poisoning |
|
|
How does lead affect the brain?
|
Can cause encephalopathy
|
|
|
How does lead affect the musculoskeletal system?
|
Drops: wrist and foot drops
|
|
|
How does lead affect the abdomen?
|
Abdominal colic
|
|
|
How do you treat lead poisoning?
|
First line treatments:
- Dimercaprol - EDTA Succimer used for chelation for kids ("it sucks to be a kid who eats lead") |
|
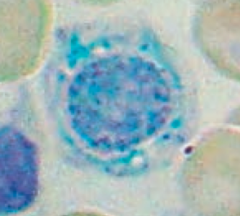
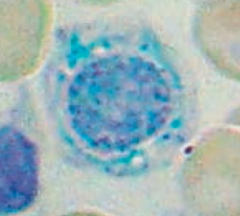
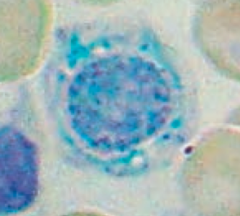
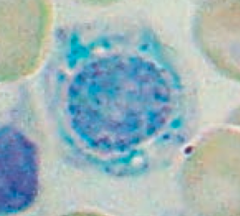
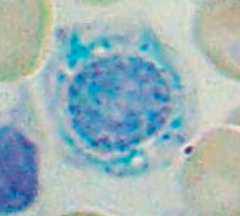
What is wrong in this picture? Cause?
|
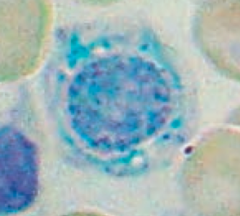
Sideroblastic Anemia
- Defect in heme synthesis - Hereditary: X-linked defect in δ-ALA synthase gene |
|
What causes sideroblastic anemia?
|
- Genetic
- Acquired (myelodysplastic syndromes) - Reversible (alcohol is most common, lead, vitamin B6 deficiency, copper deficiency, and isoniazid) |
|
What causes acquired sideroblastic anemia?
|
Myelodysplastic syndromes
|
|
|
What causes reversible sideroblastic anemia?
|
- Alcohol is most common
- Lead - Vitamin B6 deficiency - Copper deficiency - Isoniazid |
|
|
What are the lab findings in sideroblastic anemia?
|
- ↑ Iron
- Normal TIBC - ↑ Ferritin |
|
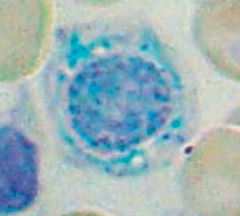
How do you treat Sideroblastic Anemia?
|
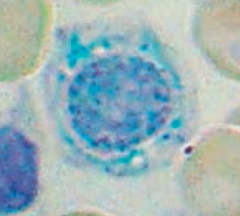
Pyridoxine (B6, cofactor for δ-ALA synthase)
|
|
|
What causes megaloblastic anemia?
|
- Impaired DNA synthesis → maturation of nucleus of precursor cells in BM delayed relative to maturation of cytoplasm
- Abnormal cell division → pancytopenia |
|
|
What are the types of macrocytic, megaloblastic anemia?
|
- Folate deficiency
- B12 deficiency (cobalamin) - Orototic aciduria |
|
|
What can cause folate deficiency?
|
- Malnutrition (eg, alcoholics)
- Malabsorption - Antifolates (eg, methotrexate, trimethoprim, phenytoin) - ↑ requirement (eg, hemolytic anemia or pregnancy) |
|
|
What are the findings of a folate deficiency?
|
- Hypersegmented neutrophils
- Glossitis - ↓ Folate, ↑ Homocysteine, but normal Methylmalonic acid * No neurologic symptoms |
|
|
What can cause B12 (Cobalamin) deficiency?
|
- Insufficient intake (eg, strict vegans)
- Malabsorption (eg, Crohn disease) - Pernicious anemia - Diphyllobothrium latum (fish tapeworm) - Proton pump inhibitors |
|
|
What are the findings of a B12 (Cobalamin) deficiency?
|
- Hypersegmented neutrophils
- Glossitis - ↓ B12, ↑ Homocysteine, ↑ Methylmalonic Acid * Neurologic symptoms: subacute combined degeneration (due to involvement of B12 in fatty acid pathways and myelin synthesis) |
|
|
What are the neurologic symptoms associated with some cases of megaloblastic anemia? Which cause?
|
Subacute combined degeneration (due to involvement of B12 in fatty acid pathways and myelin synthesis)
- Peripheral neuropathy with sensorimotor dysfunction - Dorsal columns (vibration / proprioception) - Lateral corticospinal (spasticity) - Dementia |
|
|
What causes Orotic Aciduria?
|
- Inability to convert orotic acid to UMP (de novo pyrimidine synthesis pathway) because of defect in UMP synthase
- Autosomal recessive |
|
|
How does Orotic Aciduria present?
|
Presents in children with failure to thrive and a megaloblastic anemia that cannot be cured by folate or B12
- This is because the defect is in UMP synthase, preventing the conversion of orotic acid to UMP (de novo pyrimidine synthesis pathway) |
|
|
Does Orotic Aciduria that causes megaloblastic anemia have hyperammonemia? Why?
|
No hyperammonemia
- However an ornithine transcarbamylase deficiency would cause increased orotic acid with hyperammonemia |
|
|
What are the findings of orotic aciduria?
|
- Hypersegmented neutrophils
- Glossitis - Orotic acid in urine |
|
|
How do you treat orotic aciduria?
|
Uridine monophosphate to bypass mutated enzyme
|
|
|
What can cause a non-megaloblastic macrocytic anemia? Cause?
|
Macrocytic anemia in which DNA synthesis is NOT impaired, causes:
- Liver disease - Alcoholism - Reticulocytosis → ↑ MCV - Drugs: 5-FU, Zidovudine, Hydroxyurea |
|
|
Which drugs can cause a macrocytic non-megaloblastic anemia?
|
- 5-FU
- Zidovudine - Hydroxyurea |
|
|
How do you classify normocytic, normochromic anemias?
|
Non-hemolytic or Hemolytic
- They are further classified according to the cause of the hemolysis (intrinsic vs. extrinsic to the RBC) and by the location of the hemolysis (intravascular vs. extravascular) |
|
|
In what type of anemia would you see: ↓ haptoglobin, ↑ LDH, schistocytes and ↑ reticulocytes on a peripheral blood smear, and urobilinogen in the urine? Causes?
|
Intravascular Hemolytic (Normocytic / Normochromic) Anemia
- Paroxysmal nocturnal hemoglobinuria - Mechanical destruction (aortic stenosis, prosthetic valve) - Microangiopathic hemolytic anemia |
|
|
In what type of anemia would you see: spherocytes in peripheral blood smear, ↑ LDH, and ↑ unconjugated bilirubin, which causes jaundice? Causes?
|
Extravascular Hemolytic (Normocytic / Normochromic) Anemia
- Hereditary spherocytosis |
|
|
What are the findings in intravascular hemolysis?
|
- ↓ Haptoglobin (binds and removes free hemoglobin)
- ↑ LDH - Schistocytes and ↑ reticulocytes on peripheral blood smear - Urobilinogen in urine |
|
|
What are the causes of intravascular hemolysis?
|
- Paroxysmal nocturnal hemoglobinuria
- Mechanical destruction (aortic stenosis, prosthetic valve) - Microangiopathic hemolytic anemia |
|
|
What are the findings in extravascular hemolysis?
|
- Macrophages in spleen clears RBC
- Spherocytes in peripheral smear - ↑ LDH - ↑ Unconjugated bilirubin → Jaundice |
|
|
What are the causes of intravascular hemolysis?
|
Hereditary Spherocytosis
|
|
|
What are the non-hemolytic, normocytic anemias?
|
- Anemia of chronic disease
- Aplastic anemia - Chronic kidney disease |
|
|
How can chronic diseases cause anemia? What type?
|
Non-hemolytic, normocytic anemia, but can become a microcytic, hypochromic anemia
- Chronic inflammation → ↑ Hepcidin (released by liver, binds ferroportin on intestinal mucosal cells and macrophages, inhibiting iron transport) - ↓ Release of iron from macrophages |
|
|
What are the lab findings in anemia of chronic disease?
|
- ↓ Iron
- ↓ TIBC - ↑ Ferritin (stores iron in tissues) - ↑ Hepcidin (inhibits iron transportation) |
|
|
In general, what causes aplastic anemia?
|
Failure or destruction of myeloid stem cells
|
|
|
What can cause failure or destruction of myeloid stem cells, leading to aplastic anemia?
|
- Radiation and drugs (benzene, chloramphenicol, alkylating agents, anti-metabolites)
- Viral agents (parvovirus B19, EBV, HIV, HCV) - Fanconi anemia (DNA repair defect) - Idiopathic (immune mediated, 1° stem cell defect), may follow acute hepatitis |
|
|
What drugs can cause aplastic anemia?
|
- Benzene
- Chloramphenicol - Alkylating agents - Anti-metabolites |
|
|
What viral agents can cause aplastic anemia?
|
- Parvovirus B19
- HIV - EBV - HCV |
|
|
What is wrong in Fanconi Anemia? What does it cause?
|
- DNA repair defect
- Causes aplastic anemia (non-hemolytic, normocytic) |
|
|
What are the lab findings in Aplastic Anemia?
|
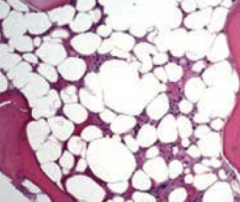
- Pancytopenia, severe anemia, leukopenia, and thrombocytopenia
- Normal cell morphology, but hypocellular bone marrow with fatty infiltration (dry BM tap) |
|
|
What are the symptoms of Aplastic Anemia?
|
- Fatigue
- Malaise - Pallor - Purpura - Mucosal bleeding - Petechiae - Infection |
|
|
How do you treat aplastic anemia?
|
- Withdrawal of offending agent
- Immunosuppressive regimens (antithymocyte globulin, cyclosporin) - Allogeneic bone marrow transplantation - RBC and platelet transfusion - G-CSF (stimulates granulocyte growth) or GM-CSF (stimulates granulocytes and monocytes) |
|
|
What immunosuppressive regimens can be used to treat aplastic anemia?
|
- Antithymocyte globulin
- Cyclosporine |
|
|
Why does chronic kidney disease lead to anemia?
|
↓ EPO → ↓ Hemoatopoiesis
|
|
|
What are the causes of intrinsic hemolytic normocytic anemia? Which are extravascular vs intravascular?
|
- Hereditary spherocytosis (E)
- G6PD deficiency (I/E) - Pyruvate kinase deficiency (E) - HbC defect (E) - Paroxysmal nocturnal hemoglobinuria (I) - Sickle cell anemia (E) |
|
|
What disease is caused by a defect in proteins interacting with the RBC membrane skeleton and plasma membrane? Which proteins?
|
Hereditary Spherocytosis
- Ankyrin - Band 3 - Protein 4.2 - Spectrin |
|
|
What are the implications of a defect in ankyrin, band 3, protein 4.2, and/or spectrin?
|
Hereditary Spherocytosis:
- Less membrane causes small and round RBCs with no central pallor - ↑ MCHC, ↑ red cell distribution width - Premature removal of RBCs by spleen |
|
|
What are the findings in patients with Hereditary Spherocytosis?
|
- Splenomegaly
- Aplastic crisis (precipitated by parvovirus B19 infection) |
|
|
What are the lab findings in Hereditary Spherocytosis?
|
- Osmotic fragility test (+)
- Eosin-5-maleimide binding test (useful for screening) - Normal to ↓ MCV with abundance of cells - Masks microcytia |
|
|
How do you treat Hereditary Spherocytosis?
|
Splenectomy because the spleen is prematurely removing the RBCs
|
|
|
What is the most common enzymatic disorder of RBCs? Implications?
|
G6PD deficiency (X-linked recessive) → ↓ glutathione → ↑ RBC susceptibility to oxidant stress → intrinsic hemolytic normocytic anemia (intravascular and extravascular)
|
|
|
What are the classic causes of oxidant stress that lead to hemolysis in patients with a G6PD deficiency?
|
- Sulfa drugs
- Anti-malarials - Infections - Fava beans |
|
|
What are the symptoms of patients with G6PD deficiency?
|
- Back pain
- Hemoglobinuria a few days after oxidant stress |
|
|
What are the labs characteristic of G6PD deficiency?
|
Blood smear shows RBCs with Heinz bodies and bite cells
|
|
|
If your patient has back pain and hemoglobinuria a few days after oxidant stress and their blood smear shows RBCs with Heinz bodies and bite cells, what diagnosis should you consider? Cause?
|
G6PD Deficiency hemolytic anemia
- X-linked recessive - ↓ Glutathione → ↑ RBC susceptibility to oxidant stress |
|
|
What can cause hemolytic anemia in a newborn?
|
Pyruvate Kinase Deficiency
- Autosomal recessive - Defective pyruvate kinase → ↓ ATP → rigid RBCs |
|
|
What are the consequences of a pyruvate kinase deficiency?
|
- Defective pyruvate kinase → ↓ ATP → rigid RBCs
- Intrinsic hemolytic normocytic anemia (extra-vascular) |
|
|
What hemoglobin defects can cause an intrinsic hemolytic normocytic anemia
|
- HbC defect
- Sickle cell anemia (HbS defect) |
|
|
What causes an HbC defect?
|
Glutamic acid to LYSINE mutation at residue 6 of β-globin
(ly-C-ine) |
|
|
What causes an HbS defect?
|
Glutamic acid to VALINE mutation at residue 6 of β-globin
|
|
|
What are the implications of a glutamic acid to lysine mutation at residue 6 in β-globin?
|
HbC defect
- Causes intrinsic hemolytic normocytic anemia (extra-vascular) - Patients with HbSC (1 of each mutant gene) have milder disease than those with HbSS genes |
|
|
What are the implications of a glutamic acid to valine mutation at residue 6 in β-globin?
|
HbS defect / Sickle Cell Anemia
- Heterozygotes (1 copy of HbS) = Sickle Cell Trait = provides resistance to malaria - Homozygotes (2 copies of HbS) = Sickle Cell Disease = more severe |
|
|
What is the pathogenesis responsible for Sickle Cell Anemia?
|
- HbS point mutation
- Low O2, dehydration, or acidosis precipitates sickling (deoxygenated HbS polymerizes) - Results in anemia and vaso-occlusive disease |
|
|
What precipitates sickling in Sickle Cell Anemia?
|
- Low O2
- Dehydration - Acidosis |
|
|
What is the effect of Sickle Cell Disease on newborns?
|
They are initially asymptomatic because of ↑ HbF and ↓ HbS
|
|
|
How common is the HbS trait? Effect?
|
8% of African Americans carry the HbS trait
- Provides resistance to malaria |
|
|
What is the appearance of Sickle Cell Anemia on a peripheral blood smear?
|
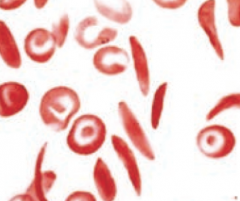
Sickled cells are crescent-shaped
|
|
|
What happens to the bones in patients with Sickle Cell Anemia?
|
"Crew cut" on skull x-ray because of BM expansion from ↑ erythropoiesis (also seen in thalassemias)
|
|
|
What are the potential complications of Sickle Cell Anemia?
|
- Aplastic crisis
- Autosplenectomy → ↑ risk of infection - Splenic sequestration crisis - Salmonella osteomyelitis - Painful crisis (vaso-occlusive) - Acute chest syndrome - Avascular necrosis - Stroke - Renal papillary necrosis |
|
|
What can cause an aplastic crisis in a patient with Sickle Cell Anemia?
|
Parvovirus B19 infection
|
|
|
What are the effects on Sickle Cell Anemia on the spleen? Complications?
|
- Autosplenectomy → formation of Howell Jolly bodies → ↑ risk of infection with encapsulated organisms
- Early splenic dysfunction occurs in childhood - Splenic sequestration crisis |
|
|
What bone infection are patients with Sickle Cell Anemia at risk for?
|
Salmonella Osteomyelitis
|
|
|
What kind of vaso-occlusive, painful crises can result from Sickle Cell Anemia?
|
- Dactylitis (painful hand swelling)
- Acute chest syndrome (most common cause of death in adults) - Avascular necrosis - Stroke |
|
|
How are the kidneys affected by Sickle Cell Anemia?
|
Renal Papillary Necrosis
- Due to low O2 in papilla - Also seen in heterozygotes Microhematuria - Due to medullary infarcts |
|
|
How do you diagnose Sickle Cell Anemia?
|
Hemoglobin electrophoresis
|
|
|
How do you treat Sickle Cell Anemia?
|
- Hydroxyurea (↑ HbF)
- Bone marrow transplantation |
|
|
What is the cause of Paroxysmal Nocturnal Hemoglobinuria?
|
↑ Complement mediated RBC Lysis:
- Impaired synthesis of GPI anchor for decay-accelerating factor that protects RBC membrane from complement - Acquired mutation in a hematopoietic stem cell |
|
|
What is there risk of in patients with Paroxysmal Nocturnal Hemoglobinuria?
|
Increased incidence of acute leukemias
|
|
|
What lab findings are there in Paroxysmal Nocturnal Hemoglobinuria?
|
- Coombs (-) Hemolytic anemia
- Pancytopenia - Venous Thrombosis - CD55 / 59 (-) RBCs on flow cytometry |
|
|
How do you treat Paroxysmal Nocturnal Hemoglobinuria?
|
Eculizumab
|
|
|
What are the types of extrinsic hemolytic normocytic anemias?
|
- Auto-immune hemolytic anemia
- Microangiopathic anemia - Macroangiopathic anemia - Infections |
|
|
What are the types of autoimmune hemolytic anemia?
|
- Warm agglutinin (IgG)
- Cold agglutinin (IgM) |
|
|
What is warm agglutinin auto-immune hemolytic anemia associated with?
|
- IgG
- Chronic anemia seen in in SLE, CLL, or with certain drugs (eg, α-methyldopa) - Can be idiopathic |
|
|
What is cold agglutinin auto-immune hemolytic anemia associated with?
|
- IgM
- Acute anemia triggered by cold, seen in CLL, Mycoplasma pneumonia infections, or infectious mononucleosis - Can be idiiopathic |
|
|
What are the lab results for patients with auto-immune hemolytic anemia?
|
Coombs test (+)
|
|
|
What are the types of Coombs tests? What do they indicate?
|
- Direct Coombs test: anti-Ig antibody (Coombs reagent) added to patient's blood, RBCs agglutinate if RBCs are coated in Ig
- Indirect Coombs test: normal RBCs added to patient's serum, if serum has anti-RBC surface Ig, RBCs agglutinate when anti-Ig antibodies (Coombs reagent) is added |
|
|
What test adds anti-Ig antibody (Coombs reagent) to patient's blood? What is a positive result?
|
Direct Coombs test:
- Positive: RBCs agglutinate, indicates RBCs are coated with Ig |
|
|
What test adds normal RBCs to the patient's serum and then adds anti-Ig antibodies (Coombs reagent)? What is a positive result?
|
Indirect Coombs test:
- Positive: RBCs agglutinate when reagent is added if serum has anti-RBC surface Ig |
|
|
What causes Microangipathic Anemia?
|
RBCs are damaged when passing through obstructed or narrowed vessel lumina
- Seen in DIC, TTP-HUS, SLE, and malignant hypertension |
|
|
What causes Macroangipathic Anemia?
|
RBCs are damaged mechanically because of prosthetic heart valves or aortic stenosis
|
|
|
What are the findings of patients with Microangipathic vs Macroangiopathic Anemia?
|
Both have schistocytes (helmet cells) on blood smear due to destruction of RBCs
Difference is based on cause |
|
|
What infections can cause an extrinsic hemolytic normocytic anemia?
|
- Malaria
- Babesia |
|
|
What type of anemia has the following lab values:
- Serum iron: ↓ - Transferrin or TIBC: ↑ - Ferritin: ↓ - % Transferrin Saturation (serum iron/TIBC): ↓↓ What is the primary change? |
Iron Deficiency Anemia
Primary change is ↓ in serum iron |
|
|
What type of anemia has the following lab values:
- Serum iron: ↓ - Transferrin or TIBC: ↓ - Ferritin: ↑ - % Transferrin Saturation (serum iron/TIBC): - What is the primary change? |
Anemia of Chronic Disease
Primary change is ↑ in ferritin |
|
|
What type of anemia has the following lab values:
- Serum iron: ↑ - Transferrin or TIBC: ↓ - Ferritin:↑ - % Transferrin Saturation (serum iron/TIBC): ↑↑ What is the primary change? |
Hemochromatosis
Primary change is ↑ in serum iron |
|
|
What type of anemia has the following lab values:
- Serum iron: - - Transferrin or TIBC: ↑ - Ferritin: - - % Transferrin Saturation (serum iron/TIBC): ↓ What is the primary change? |
Pregnancy or OCP use
Primary change is ↑ in Transferrin or TIBC (indirectly measures transferrin) |
|
|
What is the function of Transferrin?
|
Transports iron in the blood
|
|
|
What is the function of Ferritin?
|
Primary iron storage protein in body
|
|
|
Why does transferrin decrease in anemia of chronic disease?
|
Evolutionary reasoning: pathogens use circulating iron to thrive, the body has adapted a system in which iron is stored within the cells of the body and prevents pathogens from acquiring circulating iron
|
|
|
Which types of anemia have decreased transferrin (protein that transports iron in blood)?
|
- Anemia of chronic disease
- Hemochromatosis |
|
|
Which types of anemia have increased transferrin (protein that transports iron in blood)?
|
- Iron deficiency anemia
- Pregnancy / OCP use (primary change) |
|
|
Which types of anemia have decreased ferritin (1° iron storage protein in body)?
|
Iron deficiency anemia
|
|
|
Which types of anemia have increased ferritin (1° iron storage protein in body)?
|
- Anemia of chronic disease (1° change)
- Hemochromatosis |
|
|
Which types of anemia have decreased % transferrin saturation?
|
- ↓↓ in iron deficiency anemia
- ↓ in pregnancy / OCP use |
|
|
Which types of anemia have increased % transferrin saturation?
|
Hemochromatosis
|
|
|
What are the types of leukopenias?
|
- Neutropenia
- Lymphopenia - Eosinopenia |
|
|
What is the definition and possible causes of Neutropenia?
|
- Absolute neutrophil count <1500 cells / mm3
Causes: - Sepsis / post-infection - Drugs (including chemotherapy) - Aplastic anemia - SLE - Radiation |
|
|
What is the definition and possible causes of Lymphopenia?
|
- Absolute lymphocyte count <1500 cells / mm3 (<3000 cells/mm3 in children)
Causes: - HIV - DiGeorge syndrome - SCID - SLE - Corticosteroids - Radiation - Sepsis - Post-operative |
|
|
How do corticosteroids affect levels of leukocytes?
|
- Corticosteroids cause neutrophilia, but eosinopenia and lymphopenia
- Corticosteroids ↓ activation of neutrophil adhesion molecules, impairing migration out of the vasculature to sites of inflammation - In contrast, corticosteroids sequester eosinophils in lymph nodes and cause apoptosis of lymphocytes |
|
|
What are the possible causes of Eosinopenia?
|
- Cushing syndrome
- Corticosteroids |
|
|
What is the name of the hereditary or acquired conditions of defective heme synthesis? What do they lead to?
|
Porphyrias:
- Leads to accumulation of heme precursors |
|
|
How does lead affect heme synthesis?
|
Lead inhibits specific enzymes needed in heme synthesis, leading to a condition similar to porphyrias
|
|
|
What are the types of porphyrias?
|
- Acute intermittent porphyria
- Porphyria cutanea tarda |
|
|
What enzymes in heme synthesis are affected by lead poisoning?
|
- Ferrochelatase
- ALA dehydratase |
|
|
What substrates accumulate as a result of lead poisoning?
|
- Protoporphyrin
- δ-ALA (blood) |
|
|
What are the presenting symptoms of lead poisoning?
|
- Microcytic anemia
- GI and kidney disease - Children: exposure to lead paint → mental deterioration - Adults: environmental exposure (battery, ammunition, radiator factor) → headache, memory loss, demyelination |
|
|
What enzyme is affected by Acute Intermittent Porphyria?
|
Porphobilinogen Deaminase
|
|
|
What substrates accumulate as a result of Acute Intermittent Porphyria?
|
- Porphobilinogen
- δ-ALA - Corporphobilinogen (urine) |
|
|
What are the presenting symptoms of Acute Intermittent Porphyria?
|
Symptoms 5 P's:
- Painful abdomen - Port wine-colored urine - Polyneuropathy - Psychological disturbances - Precipitated by drugs, alcohol and starvation |
|
|
What can cause presentation of symptoms of Acute Intermittent Porphyria?
|
- Drugs
- Alcohol - Starvation |
|
|
What enzyme is affected by Porphyria Cutanea Tarda?
|
Uroporphyrinogen Decarboxylase
|
|
|
What substrates accumulate as a result of Porphyria Cutanea Tarda?
|
Uroporphyrin (tea-colored urine)
|
|
|
What are the presenting symptoms of Porphyria Cutanea Tarda?
|
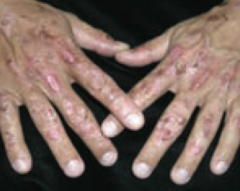
- Blistering cutaneous photosensitivity
- Tea-colored urine (due to uroporphyrin) |
|
|
What disease is caused by a defective Ferrochelatase and ALA dehydratase? What accumulates and what are the presenting symptoms?
|
Lead Poisoning
- Accumulation of protoporphyrin and δ-ALA (blood) - Microcytic anemia - GI and kidney disease - Children: exposure to lead paint → mental deterioration - Adults: environmental exposure (battery, ammunition, radiator factor) → headache, memory loss, demyelination |
|
|
What disease is caused by a defective Porphobilinogen Deaminase? What accumulates and what are the presenting symptoms?
|
Acute Intermittent Porphyria
- Accumulation of Porphobilinogen, δ-ALA, and Coporphobilinogen (urine) Symptoms 5 P's: - Painful abdomen - Port wine-colored urine - Polyneuropathy - Psychological disturbances - Precipitated by drugs, alcohol and starvation |
|
|
How do you treat Acute Intermittent Porphyria?
|
Glucose and Heme, which inhibit ALA synthase
|
|
|
What disease is caused by a defective Uroporphyrinogen Decarboxylase? What accumulates and what are the presenting symptoms?
|
Porphyria Cutanea Tarda
- Accumulation of uroporphyrin (tea-colored urine) - Blistering cutaneous photosensitivity |
|
|
What is the most common porphyria?
|
Porphyria Cutanea Tarda
- Defect in uroporphyrinogen decarboxylase - Accumulation of uroporphyrin (tea-colored urine) - Blistering cutaneous photosensitivity |
|
|
What enzyme is defective in sideroblastic anemia?
|
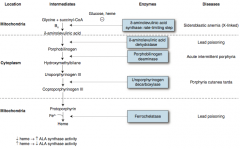
δ-Aminolevulinic Acid Synthase (rate-limiting step of heme synthesis
- X-linked |
|
|
How does the level of heme affect ALA synthase activity?
|
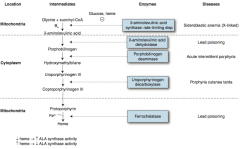
↓ Heme → ↑ ALA Synthase activity
↑ Heme → ↓ ALA Synthase activity (ALA Synthase is the rate-limiting step in heme synthesis) |
|
|
What tests can be used to assess for coagulation disorders? What do they test?
|
- PT test - function of common and EXTRINSIC pathway (factors I, II, V, VII, and X)
- PTT test - function of common and INTRINSIC pathway (all factors except VII, and XIII) |
|
|
Defects in what factors can cause an increased PT time?
|
Factors I, II, V, VII, and X
|
|
|
Defects in what factors can cause an increased PTT time?
|
All factors except VII and XIII
|
|
|
Which disorders cause a normal PT but a prolonged PTT?
|
Hemophilia A and B
- Hemophilia A: deficiency of Factor VIII - Hemophilia B: deficiency of Factor IX Affects intrinsic pathway |
|
|
What are the signs / symptoms associated with hemophilia A or B?
|
- Prolonged PTT but normal PT
- Macrohemorrhage in hemophilia: hemarthroses (bleeding into joints), easy bruising |
|
|
How do you treat Hemophilia A or B?
|
Hemophilia A: treat with recombinant factor VIII
|
|
|
Which disorders cause a prolonged PT and PTT?
|
Vitamin K deficiency
- ↓ Synthesis of factors II, VII, IX, X, protein C, and protein S |
|
|
What are the signs / symptoms of a Vitamin K deficiency?
|
- Prolonged PT and PTT times
- Normal bleeding time - General coagulation defect |
|
|
What are the types of platelet disorders?
|
- Bernard-Soulier Syndrome
- Glanzmann Thromboasthenia - Immune Thrombocytopenia (ITP) - Thrombotic Thrombocytopenic Purpura (TTP) |
|
|
What common findings are associated with defects in platelet plug formation?
|
Increased bleeding time (BT)
|
|
|
What are the signs / symptoms of platelet abnormalities?
|
- Microhemorrhage: mucus membrane bleeding, epistaxis, petechiae, purpura
- ↑ bleeding time - Possible ↓ platelet count (PC) |
|
|
Which platelet disorders have decreased platelet counts?
|
- Bernard-Soulier Syndrome
- Immune Thrombocytopenia (ITP) - Thrombotic Thrombocytopenic Purpura (TTP) Normal platelet count: - Glanzmann Thromboasthenia |
|
|
What disorder is caused by decreased GpIb? Mechanism / effect?
|
Bernard-Soulier Syndrome
- ↓ GpIb → defect in platelet to vWF adhesion - Leads to defect in platelet plug formation - ↓ Platelet count and ↑ Bleeding time |
|
|
What disorder is caused by decreased GpIIb/IIIa? Mechanism / effect?
|
Glanzmann Thrombasthenia
- ↓ GpIIb/IIIa → defect in platelet-to-platelet aggregation - Defect in platelet plug formation - Normal platelet count with ↑ Bleeding time - Labs: blood smear shows NO platelet clumping |
|
|
What disorder is caused by anti-GpIIb/IIIa antibodies? Mechanism / effect?
|
Immune Thrombocytopenia
- Anti-GpIIb/IIIa antibodies → splenic macrophage consumption of platelet / antibody complex - May be triggered by a viral illness - ↓ Platelet survival → ↓ Platelet count and ↑ Bleeding time - Labs: ↑ Megakaryocytes on BM biopsy |
|
|
What disorder is caused by inhibition or deficiency of ADAMTS 13? Mechanism / effect?
|
Thrombotic Thrombocytopenic Purpura
- Inhibition or deficiency of ADAMTS 13 (vWF metalloprotease) → ↓ degradation of vWF multimers - ↑ Large vWF multimers → ↑ platelet adhesion → ↑ platelet aggregation and thrombosis - ↓ Platelet survival → ↓ platelet cut and ↑ bleeding time - Labs: schistocytes and ↑ LDH |
|
|
What are the symptoms of Thrombotic Thrombocytopenic Purpura?
|
Pentad of neurologic and renal symptoms, fever, thrombocytopenia, and microangiopathic hemolytic anemia
|
|
|
How do you treat Thrombotic Thrombocytopenic Purpura?
|
Exchange transfusion and steroids
|
|
|
What is the mechanism of Bernard-Soulier Syndrome?
|
↓ GpIb → defect in platelet to vWF adhesion
- Defect in platelet plug formation |
|
|
What is the mechanism of Glanzmann Thrombasthenia?
|
↓ GpIIb/IIIa → defect in platelet to platelet aggregation
- Defect in platelet plug formation - Blood smear shows no platelet clumping |
|
|
What is the mechanism of Immune Thrombocytopenia?
|
Anti-GpIIb/IIIa antibodies → splenic macrophage consumption of platelet / antibody complex
- May be triggered by a viral illness - ↓ Platelet survival - ↑ Megakaryocytes on BM biopsy |
|
|
Which platelet disorder shows ↑ Megakaryocytes on BM biopsy?
|
Immune Thrombocytopenia
- Anti-GpIIb/IIIa antibodies → splenic macrophage consumption of platelet / antibody complex |
|
|
What is the mechanism of Thrombotic Thrombocytopenic Purpura?
|
Inhibition or deficiency of ADAMTS 13 (vWF metalloprotease) → ↓ degradation of vWF multimres
Leads to ↑ large vWF multimers → ↑ platelet adhesion → ↑ platelet aggregation and thrombosis ↓ Platelet survival |
|
|
Which platelet disorder has schistocytes?
|
Thrombotic Thrombocytopenic Purpura because of increased platelet aggregation and thrombosis that causes microangiopathic hemolytic anemia
|
|
|
What are the mixed platelet and coagulation disorders?
|
- Von Willebrand Disease
- DIC |
|
|
Which disorder causes a prolonged bleeding time, a normal or prolonged PTT, and normal platelet count and PT?
|
von Willebrand Disease
|
|
|
Which disorder causes a prolonged bleeding time, PT, PTT, and decreased platelet count?
|
DIC
|
|
|
What is the mechanism of pathogenesis of von Willebrand Disease?
|
- Intrinsic pathway coagulation defect: ↓ vWF → normal or ↑ PTT (depends on severity; vWF acts to carry / protect factor VIII)
- Defect in platelet plug formation: ↓ vWF → defect in platelet-to-vWF adhesion |
|
|
How can von Willebrand Disease cause a prolonged PTT?
|
vWF acts to carry / protect factor VIII
|
|
|
What is the most common inherited bleeding disorder? How is it inherited?
|
von Willebrand Disease - autosomal dominant
|
|
|
How do you diagnose von Willebrand disease?
|
Ristocetin cofactor assay (↓ agglutination is diagnostic)
|
|
|
How do you treat von Willebrand disease?
|
DDAVP, which releases vWF stored in endothelium
|
|
|
What is the mechanism of pathogenesis of DIC?
|
Widespread activation of clotting leads to a deficiency in clotting factors, which creates a bleeding state
|
|
|
What are the possible causes of DIC?
|
STOP Making New Thrombi:
- Sepsis (G-) - Trauma - Obstetric copmlications - Pancreatitis (acute) - Malignancy - Nephrotic syndrome - Transfusion |
|
|
What labs are indicative of DIC?
|
- Schistocytes on peripheral blood smear
- ↑ Fibrin split products (D-dimers) - ↓ Fibrinogen - ↓ Factors V and VIII |
|
|
What are the hereditary thrombosis syndromes that lead to hypercoagulability?
|
- Factor V Leiden
- Prothrombin gene mutation - Antithrombin deficiency - Protein C or S deficiency |
|
|
What is wrong in Factor V Leiden? Implications?
|
- Production of mutant factor V that is resistant to degradation by activated Protein C
- Leads to hypercoagulability and thrombosis |
|
|
What is the most common cause of inherited hypercoagulability in whites?
|
Factor V Leiden
- Production of mutant factor V that is resistant to degradation by activated Protein C |
|
|
What is wrong with a Prothrombin Gene Mutation? Implications?
|
- Mutation in 3' untranslated region → ↑ production of prothrombin → ↑ plasma levels and venous clots
- Leads to hypercoagulability and thrombosis |
|
|
What is wrong in Anti-Thrombin Deficiency? Implications?
|
- Inherited deficiency of antithrombin, no direct effect on PT, PTT, or thrombin time
- Diminishes the increase in PTT following heparin administration - Can also be acquired: renal failure / nephrotic syndrome → antithrombin loss in urine → ↑ factors II and X |
|
|
What renal pathology can cause a hypercoagulable state?
|
Renal failure / nephrotic syndrome → antithrombin loss in urine → ↑ factors II and X
Leads to hypercoagulability and increased risk for thrombosis |
|
|
What is wrong in Protein C or S deficiency? Implications?
|
- ↓ Ability to inactivate factors V and VIII
- ↑ Risk of thrombotic skin necrosis with hemorrhage following administration of warfarin |
|
|
What diagnosis should you think of if your patient develops skin and subcutaneous necrosis after administering warfarin?
|
Protein C deficiency:
Protein C Cancels Coagulation |
|
|
What are the forms of blood transfusion therapy?
|
- Packed RBCs
- Platelets - Fresh frozen plasma - Cryoprecipitate |
|
|
What form of blood transfusion therapy would you give to a patient with acute blood loss? Effect?
|
Packed RBCs:
- ↑ Hb and O2 carrying capacity |
|
|
What form of blood transfusion therapy would you give to a patient with severe anemia? Effect?
|
Packed RBCs:
- ↑ Hb and O2 carrying capacity |
|
|
What form of blood transfusion therapy would you give to a patient to stop significant bleeding (thrombocytopenia, qualitative platelet defects)? Effect?
|
Platelets:
- ↑ platelet count (↑ ~ 5000/mm3/unit) |
|
|
What form of blood transfusion therapy would you give to a patient with DIC? Effect?
|
Fresh Frozen Plasma:
- ↑ coagulation factor levels |
|
|
What form of blood transfusion therapy would you give to a patient with cirrhosis? Effect?
|
Fresh Frozen Plasma:
- ↑ coagulation factor levels |
|
|
What form of blood transfusion therapy would you give to a patient with a warfarin overdose? Effect?
|
Fresh Frozen Plasma:
- ↑ coagulation factor levels |
|
|
What form of blood transfusion therapy would you give to a patient with TTP/HUS? Effect?
|
Exchange transfusion of Fresh Frozen Plasma
- ↑ coagulation factor levels |
|
|
What form of blood transfusion therapy would you give to a patient with coagulation factor deficiencies involving fibrinogen and factor VIII? Effect?
|
Cryoprecipitate
- Contains fibrinogen, factor VIII, factor XIII, vWF, and fibronectin |
|
|
What are the risks of a blood transfusion?
|
- Infection transmission (low)
- Transfusion reactions - Iron overload - Hypocalcemia (citrate is a calcium chelator) - Hyperkalemia (RBCs may lyse in old blood units) |
|
|
What are the clinical uses of packed RBCs? Dosage effect?
|
- Use for acute blood loss or severe anemia
- ↑ Hb and O2 carrying capacity |
|
|
What are the clinical uses of platelet transfusions? Dosage effect?
|
- Use to stop significant bleeding (thrombocytopenia or qualitative platelet defects)
- ↑ platelet count (↑ ~ 5000/mm3/unit) |
|
|
What are the clinical uses of fresh frozen plasma transfusions? Dosage effect?
|
- Use for DIC, cirrhosis, warfarin overdose, exchange transfusion in TTP/HUS
- ↑ coagulation factor levels |
|
|
What are the clinical uses of cryoprecipitate? Dosage effect?
|
- Treat coagulation factor deficiencies involving fibrinogen and factor VIII
- Contains fibrinogen, factor VIII, factor XIII, vWF, and fibronectin |