Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
277 Cards in this Set
- Front
- Back
|
What are the sites of metabolism? |
- Mitochondria
- Cytoplasm - Both |
|
|
Which processes take place in the mitochondria?
|
- Fatty acid oxidation (β-oxidation)
- Acetyl-CoA production - TCA cycle - Oxidative Phosphorylation |
|
|
Which processes take place in the cytosol?
|
- Glycolysis
- Fatty acid synthesis - HMP shunt - Protein synthesis (RER) - Steroid synthesis (SER) - Cholesterol synthesis |
|
|
Which processes take place in the mitochondria and cytosol?
|
- Heme synthesis
- Urea cycle - Gluconeogenesis HUGs take TWO (ie, both) |
|
|
What is the action of a "kinase"?
|
Uses ATP to add high-energy phosphate group onto substrate (eg, phosphofructokinase)
|
|
|
What is the action of a "phosphorylase"?
|
Adds inorganic phosphate onto substrate WITHOUT using ATP (eg, glycogen phosphorylase)
|
|
|
What is the action of a "phosphatase"?
|
Removes phosphate group from substrate (eg, fructose-1,6-bisphosphatase)
|
|
|
What is the action of a "dehydrogenase"?
|
Catalyzes oxidation-reduction reactions (eg, pyruvate dehydrogenase)
|
|
|
What is the action of a "carboxylase"?
|
Transfers CO2 groups with the help of biotin (eg, pyruvate carboxylase)
|
|
|
What is the rate determining enzyme and regulators of glycolysis?
|
- Phosphofructokinase-1 (PFK-1)
- AMP (+) - Fructose-2,6-BP (+) - ATP (-) - Citrate (-) |
|
|
What is the rate determining enzyme and regulators of gluconeogenesis?
|
- Fructose-1,6-bisphosphatase
- ATP (+) - AMP (-) - Fructose-2,6-BP (-) |
|
|
What is the rate determining enzyme and regulators of the TCA cycle?
|
- Isocitrate Dehydrogenase
- ADP (+) - ATP (-) - NADH (-) |
|
|
What is the rate determining enzyme and regulators of glycogen synthesis?
|
- Glycogen synthase
- Glucose (+) - Insulin (+) - Epinephrine (-) - Glucagon (-) |
|
|
What is the rate determining enzyme and regulators of glycogenolysis?
|
- Glycogen phosphorylase
- AMP (+) - Epinephrine (+) - Glucagon (+) - Insulin (-) - ATP (-) |
|
|
What is the rate determining enzyme and regulators of HMP Shunt?
|
- Glucose-6-Phosphate Dehydrogenase (G6PD)
- NADP+ (+) - NADPH (-) |
|
|
What is the rate determining enzyme and regulators of de novo pyrimidine synthesis?
|
Carbamoyl phosphate synthetase II
(I is for the urea cycle) |
|
|
What is the rate determining enzyme and regulators of de novo purine synthesis?
|
- Glutamine-PRPP amidotransferase
- AMP (-) - IMP (-) - GMP (-) |
|
|
What is the rate determining enzyme and regulators of the urea cycle?
|
- Carbamoyl phosphate synthetase I
(II is for de novo pyrimidine synthesis) - N-acetylglutamate (+) - |
|
|
What is the rate determining enzyme and regulators of fatty acid synthesis?
|
- Acetyl-CoA carboxylase (ACC)
- Insulin (+) - Citrate (+) - Glucagon (-) - Palmitoyl-CoA (-) |
|
|
What is the rate determining enzyme and regulators of fatty acid oxidation?
|
- Carnitine acyltransferase I
- Malonyl-CoA (-) |
|
|
What is the rate determining enzyme and regulators of ketogenesis?
|
HMG-CoA Synthase
(reductase for cholesterol synthesis) |
|
|
What is the rate determining enzyme and regulators of cholesterol synthesis?
|
- HMG-Coa Reductase
(synthase for ketogenesis) - Insulin (+) - Thyroxine (+) - Glucagon (-) - Cholesterol (-) |
|
|
What is the net gain of aerobic metabolism of glucose?
|
Malate-Aspartate shuttle (heart and liver):
- Produces 32 ATP Glycerol-3-Phosphate shuttle (muscle): - Produces 30 ATP |
|
|
What is the net gain of anaerobic metabolism of glucose?
|
Anaerobic glycolysis:
- 2 net ATP / glucose molecule |
|
|
What is the purpose of ATP?
|
ATP hydrolysis can be coupled to energetically unfavorable reactions
|
|
|
Which molecule carries phosphoryl groups in its activated form?
|
ATP
|
|
|
Which molecule carries electrons in its activated form?
|
- NADH
- NADPH - FADH2 |
|
|
Which molecule carries Acyl groups in its activated form?
|
- Coenzyme A
- Lipoamide |
|
|
Which molecule carries CO2 in its activated form?
|
Biotin
|
|
|
Which molecule carries 1-C units in its activated form?
|
THF
|
|
|
Which molecule carries CH3 groups in its activated form?
|
SAM (S-adenosyl-methionine)
|
|
|
Which molecule carries aldehydes in its activated form?
|
TPP
|
|
|
What are the universal electron acceptors?
|
- Nicotinamides (NAD+ from vitamin B3, NADP+)
- Flavin nucleotides (FAD+ from vitamin B2) |
|
|
What is the typical function of NAD+?
|
Used in catabolic processes to carry reducing equivalents away as NADH
|
|
|
What is the typical function of NADPH?
|
- Used in anabolic processes (steroid and fatty acid synthesis) as a supply of reducing equivalents
- Respiratory burst - P-450 - Glutathione reductase |
|
|
How is NADPH produced?
|
HMP Shunt
|
|
|
What is the first step of glycolysis?
|
Phosphorylation of glucose → glucose-6-phosphate
- Hexokinase or Glucokinase depending on the tissue |
|
|
What is the action and location of Hexokinase vs Glucokinase?
|
Hexokinase
- Ubiquitous (everywhere) - High affinity (↓ Km) - Low capacity (↓ Vmax) - Uninduced by insulin - Feedback inhibition by glucose-6P (product) Glucokinase: - Liver and β cells of pancreas - Low affinity (↑ Km) - High capacity (↑ Vmax) - GLUcokinase is a GLUtton, it has a high Vmax because it cannot be satisfied - Induced by insulin - Feedback inhibition by fructose-6P |
|
|
How do Hexokinase and Glucokinase compare in location?
|
- Hexokinase: ubiquitous (everywhere)
- Glucokinase: liver and β cells of pancreas |
|
|
How do Hexokinase and Glucokinase compare in affinity for glucose?
|
- Hexokinase: high affinity (↓ Km)
- Glucokinase: low affinity (↑ Km) |
|
|
How do Hexokinase and Glucokinase compare in capacity / Vmax?
|
- Hexokinase: low capacity (↓ Vmax)
- Glucokinase: high capacity (↑ Vmax) * GLUcokinase is a GLUtton, it has a high Vmax because it cannot be satisfied |
|
|
How do Hexokinase and Glucokinase compare in their response to insulin?
|
- Hexokinase: uninduced by insulin
- Glucokinase: induced by insulin |
|
|
What happens to Hexokinase / Glucokinase at low glucose concentrations?
|
Hexokinase sequesters glucose in the tissue at high glucose concentrations
|
|
|
What happens to Hexokinase / Glucokinase at high glucose concentrations?
|
Excess glucose is stored in the liver (phosphorylation of glucose by hexokinase or glucokinase is the first step of glycogen synthesis in liver)
|
|
|
What is the net effect of glycolysis?
|
Glucose + 2 Pi + 2 ADP + 2 NAD+ →
2 Pyruvate + 2 ATP + 2 NADH + 2 H+ + 2 H2O |
|
|
How many ATP / NADH are produced by glycolysis of a single glucose molecule?
|
- 2 ATP
- 2 NADH |
|
|
Which steps of glycolyis require ATP?
|
- Glucose → Glucose-6P (by hexokinase/ubiquitous or glucokinase/liver)
- Fructose-6P → Fructose-1,6-BP (by phosphofructokinase-1) = rate-limiting step |
|
|
Which steps of glycolyis produce ATP?
|
Each produce 2 for 1 molecule of glucose:
- 1,3-BPG → 3-PG (by phosphoglycerate kinase) - Phosphoenolpyruvate (PEP) → Pyruvate (by pyruvate kinase) |
|
|
What regulates hexokinaae and glucokinase (1st step that requires ATP)?
|
- Hexokinase: inhibited by glucose-6P
- Glucokinase: inhibited by fructose-6P |
|
|
What regulates phosphofructokinase-1 (2nd step that requires ATP)?
|
Inhibited by:
- ATP - Citrate Stimulated by: - AMP - Fructose-2,6-BP |
|
|
What regulates pyruvate kinase (step that produces ATP)?
|
Inhibited by:
- ATP - Alanine Stimulated by: Fructose-1,6-BP |
|
|
How is sugar metabolism regulated during the FASTING state?
|
- ↑ Glucagon →
- ↑ cAMP → - ↑ Protein Kinase A → - ↑ FBPase-2 (Fructose Bisphosphatase-2), - ↓ PFK-2 (Phosphofructokinase-2), LESS GLYCOLYSIS |
|
|
How is sugar metabolism regulated during the FED state?
|
- ↑ Insulin →
- ↓ cAMP → - ↓ Protein Kinase A → - ↓ FBPase-2 (Fructose Bisphosphatase-2), - ↑ PFK-2 (Phosphofructokinase-2), MORE GLYCOLYSIS |
|
|
Which enzyme involved in sugar metabolism is activated by ↑ PKA? Implications?
|
FBPase-2 (Fructose Bisphosphatase-2)
- Converts Fructose-2,6-Bisphosphate → Fructose-6-Phosphate Occurs during fasting state: - ↓ GLYCOLYSIS - ↑ GLUCONEOGENESIS |
|
|
Which enzyme involved in sugar metabolism is active when there is ↓ PKA? Implications?
|
PFK-2 (Phosphofructokinase-2)
- Converts Fructose-6-Phosphate → Fructose-2,6-Bisphosphate Occurs during fed state: - ↑ GLYCOLYSIS - ↓ GLUCONEOGENESIS |
|
|
What reaction is mediated by the Pyruvate Dehydrogenase Complex?
|
Pyruvate + NAD+ + CoA →
Acetyl-CoA + CO2 + NADH |
|
|
What are the requirements of the Pyruvate Dehydrogenase Complex? How many enzymes are part of the complex?
|
Contains 3 enzymes that require 5 cofactors:
1) Pyrophosphate (B1, thiamine; TPP) 2) FAD (B2, riboflavin) 3) NAD (B3, niacin) 4) CoA (B5, pantothenic acid) 5) Lipoic Acid |
|
|
What activates the Pyruvate Dehydrogenase Complex?
|
- ↑ NAD+/NADH ratio
- ↑ ADP - ↑ Ca2+ |
|
|
What is the Pyruvate Dehydrogenase Complex similar to?
|
α-Ketoglutarate Dehydrogenase Complex
- Same cofactors, similar substrate and action - Converts α-Ketoglutarate → Succinyl-CoA (TCA Cycle) |
|
|
Which poison affects the Pyruvate Dehydrogenase Complex? Symptoms?
|
Arsenic: inhibits lipoic acid which is a necessary cofactor for the complex
Symptoms: - Vomiting - Rice water stools - Garlic breath |
|
|
What are the implications of a Pyruvate Dehydrogenase Complex Deficiency?
|
Causes backup of substrate (pyruvate and alanine) leading to LACTIC ACIDOSIS
|
|
|
What is the most common cause of a Pyruvate Dehydrogenase Complex deficiency?
|
Mutations in X-linked gene for E1-α subunit of PDC
|
|
|
What are the findings of a Pyruvate Dehydrogenase Complex deficiency (most cases d/t mutations in X-linked gene for E1-α subunit of PDC)? How do you treat?
|
Symptoms:
- Neurologic defects, usually starting in infancy Treatment: - ↑ Intake of ketogenic nutrients (eg, high fat content or ↑ lysine and leucine) - Lysine and leucine are the onLy pureLy ketogenic amino acids |
|
|
What are the possible pyruvate metabolic pathways
|
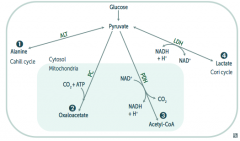
1. Pyruvate Dehydrogenase Complex: transition from glycolysis to TCA cycle
2. Alanine Aminotransferase 3. Pyruvate Carboxylase 4. Lactic Acid Dehydrogenase |
|
|
What enzyme(s) shuttle pyruvate into the TCA cycle? Cofactors?
|
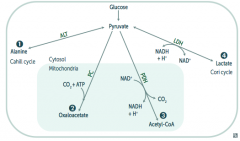
Pyruvate Dehydrogenase Complex
- Pyruvate → Acetyl-CoA - B1 - Pyrophosphate - B2 - FAD - B3 - NAD - B5 - CoA - Lipolic Acid |
|
|
What enzyme(s) converts pyruvate into a form that can be carried from the liver to muscle? Cofactors?
|
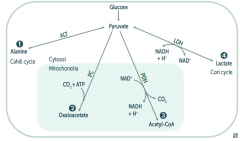
Alanine Aminotransferase
- Pyruvate → Alanine (alanine carries amino groups to the liver from muscle) - B6 - Pyridoxine |
|
|
What enzyme(s) converts pyruvate into a form that can replenish the TCA cycle or be used in gluconeogenesis? Cofactors?
|
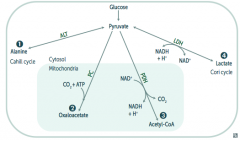
Pyruvate Carboxylase
- Pyruvate → Oxaloacetate (via addition of CO2 + ATP) - B7 - Biotin |
|
|
What enzyme(s) converts pyruvate into a form that can enter the Cori cycle? Cofactors?
|
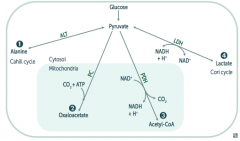
Lactic Acid Dehydrogenase
- Pyruvate → Lactate (enters Cori cycle) - B3 - NAD |
|
|
In what tissues is Lactic Acid Dehydrogenase conversion of Pyruvate to Lactate the major pathway?
|
- RBCs
- Leukocytes - Kidney medulla - Lens - Testes - Cornea |
|
|
What is the effect / results of the TCA Cycle?
|
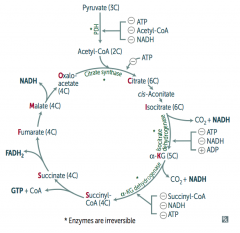
Pyruvate → Acetyl-CoA produces 1 NADH, 2 CO2
For every Acetyl-CoA - 3 NADH - 1 FADH2 - 2 CO2 - 1 GTP - 10 ATP (2x everything per glucose) |
|
|
How do you remember the substrates / intermediates of the Kreb's Cycle?
|
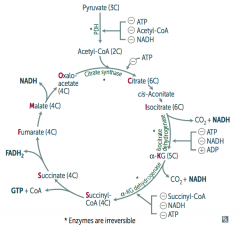
Citrate is Kreb's Starting Substrate For Making Oxaloacetate
- Citrate (6C) - Isocitrate (6C) - α-Ketoglutarate (5C) + CO2 - Succinyl-CoA (4C) + CO2 - Succinate (4C) - Fumarate (4C) - Malate (4C) - Oxaloacetate (4C) + Acetyl-CoA (2C) |
|
|
What regulates the TCA cycle?
|
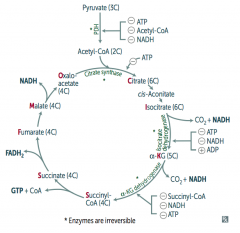
Pyruvate Dehydrogenase Complex
- ATP (-) - Acetyl-CoA (-) - NADH (-) Citrate Synthase (Oxaloacetate + Acetyl-Coa → Citrate) - ATP (-) Isocitrate Dehydrogenase (Isocitrate → α-Ketoglutarate + CO2) - ATP (-) - NADH (-) - ADP (+) α-Ketoglutarate Dehydrogenase (α-KG → Succinyl-Coa + CO2) - Succinyl-CoA (-) - NADH (-) - ATP (-) |
|
|
Which enzymes in the TCA cycle are irreversible?
|
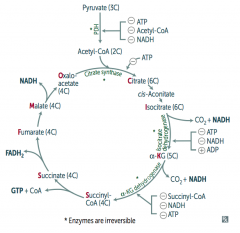
The same steps that were regulated by ATP, etc)
- Pyruvate Dehydrogenase Complex - Citrate Synthase - Isocitrate Dehydrogenase - α-Ketoglutarate Dehydrogenase |
|
|
What happens in the Electron Transport Chain and Oxidative Phosphorylation?
|
- NADH electrons from glycolysis enter mitochondria via malate-aspartate or glycerol-3-phosphate shuttle
- FADH2 electrons are transferred to complex II (at lower energy level than NADH) - Passage of e- results in formation of a H+ gradient that, coupled to oxidative phosphorylation, drives production of ATP |
|
|
How does NADH enter the Electron Transport Chain?
|
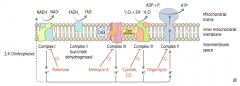
NADH electrons from glycolysis enter mitochondria via malate-aspartate or glycerol-3-phosphate shuttle
|
|
|
How does FADH2 enter the Electron Transport Chain?
|
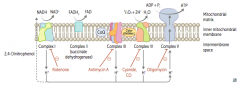
FADH2 electrons are transferred to complex II (at lower energy level than NADH)
|
|
|
How many ATP are produced per NADH and FADH2 through the Electron Transport Chain and Oxidative Phosphorylation?
|
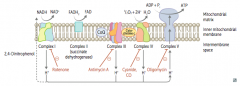
1 NADH → 2.5 ATP (remember, niacin is vitamin B3)
1 FADH2 → 1.5 ATP (remember, flavin is vitamin B2) |
|
|
What drugs / poisons can affect the Electron Transport Chain and Oxidative Phosphorylation?
|
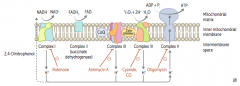
- Electron Transport Inhibitors: Rotenone, Cyanide, Antimycin A, CO
- ATP Synthase Inhibitors: Oligomycin - Uncoupling Agents: 2,4-DNP, Aspirin, Thermogenin (in brown fat) |
|
|
What is the effect of electron transport inhibitors? Which drugs?
|
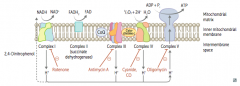
- Directly inhibits electron transport, causing a ↓ proton gradient and block of ATP synthesis
- Rotenone: inhibits Complex I - Antimycin A: inhibits Complex III - Cyanide and CO: inhibit Complex IV |
|
|
What is the effect of ATP Synthase Inhibitors? Which drugs?
|
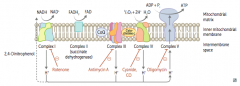
- Directly inhibit mitochondrial ATP synthase, causing an ↑ proton gradient
- No ATP is produced because electron transport stops - Oligomycin: inhibits Complex V (ADP + Pi → ATP + H2O) |
|
|
What is the effect of Uncoupling Agents? Which drugs?
|
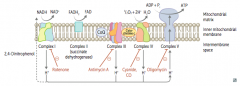
- ↑ Permeability of membrane, causing a ↓ proton gradient and ↑ O2 consumption
- ATP synthesis stops, but electron transport continues - PRODUCES HEAT - 2,4-DNP - Aspirin (fevers often occur after aspirin overdose) - Thermogenin in brown fat |
|
|
What are the irreversible enzymes in Gluconeogenesis?
|
Pathway Produces Fresh Glucose
- Pyruvate carboxylase - PEP carboxykinase - Fructose-1,6-bisphosphatase - Glucose-6-phosphatase |
|
|
What is the action of Pyruvate Carboxylase? Requirements? Location?
|
- Irreversible conversion of Pyruvate → Oxaloacetate
- Requires biotin and ATP - Activated by acetyl-CoA - In mitochondria |
|
|
What is the action of PEP Carboxykinase? Requirements? Location?
|
- Irreversible conversion of Oxaloacetate → Phosphoenolpyruvate (PEP)
- Requires GTP - In cytosol |
|
|
What is the action of Fructose-1,6-Bisphosphatase? Location?
|
- Irreversible conversion of Fructose-1,6-Bisphosphate → Fructose-6P
- In cytosol |
|
|
What is the action of Glucose-6-Phosphatase? Location?
|
- Irreversible conversion of Glucose-6-Phosphate → Glucose
- In ER |
|
|
Where does gluconeogenesis occur? Where can't it occur?
|
* Primarily in the liver
- Enzymes also found in kidney and intestinal epithelium - Muscle cannot participate in gluconeogenesis because it lacks glucose-6P |
|
|
What are the implications of a deficiency of the gluconeogenic enzymes?
|
Hypoglycemia
|
|
|
Which fatty acids can be used for gluconeogenesis? Why/why not?
|
- Odd-chain FA yield 1 propionyl-CoA during metabolism, which can enter the TCA cycle as succinyl-CoA, undergo gluconeogenesis, and serve as a glucose source
- Even-chain FA can't produce new glucose, since they yield only acetyl-CoA equivalents |
|
|
What is the function of the HMP shunt (Pentose Phosphate Pathway)?
|
- Provides a source of NADPH from an abundantly available glucose-6P
- NADPH is required for reductive reactions (eg, glutathione reduction inside RBCs) - Also yields Ribose for nucleotide synthesis and glycolytic intermediates |
|
|
How many ATP are used/gained from HMP shunt (Pentose Phosphate Pathway)?
|
No ATP are used or produced
|
|
|
Where does the HMP shunt (Pentose Phosphate Pathway) take place?
|
- Lactating mammary glands
- Liver - Adrenal cortex (sites of FA or steroid synthesis) - RBCs |
|
|
What are the reactions in the HMP shunt (Pentose Phosphate Pathway)?
|
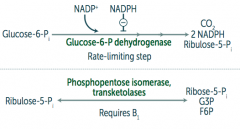
1. Oxidative (irreversible)
Glucose-6P → Ribulose-5P + CO2 + 2 NADPH by Glucose-6P dehydrogenase 2. Non-Oxidative (reversible) Ribulose-5P → Ribose-5P + G3P + F6P by Phosphopentose isomerase and transketolases |
|
|
What is the rate-limiting step of the HMP shunt (Pentose Phosphate Pathway)?
|
Glucose-6P Dehydrogenase (oxidative/irreversible reaction)
- Glucose-6P → Ribulose-5P + CO2 + 2 NADPH |
|
|
What mediates the respiratory / oxidative burst in neutrophils and/or monocytes?
|
Activation of membrane-bound NADPH oxidase (in neutrophils and monocytes)
|
|
|
What is the function of the respiratory / oxidative burst in neutrophils and/or monocytes?
|
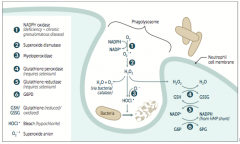
Important in the immune response → rapid release of reactive oxygen intermediates (ROIs)
* NADPH plays a role in the creation of ROIs and in their neutralization |
|
|
What do the WBCs of patients with Chronic Granulomatous Disease do? Implications for further infection?
|
- WBCs can utilize H2O2 generated by invading organisms and convert it to ROIs
- Patients are at ↑ risk for infection by catalase-positive species (eg, S. aureus and Aspergillus) because they neutralize there own H2O2, leaving WBCs without ROIs for fighting infections |
|
|
What is the function of reduced glutathione? How does glutathione get reduced?
|
- Reduced glutathione detoxifies free radicals and preoxides
- NADPH is necessary to keep glutathione reduced |
|
|
What are the implications of reduced availability of NADPH?
|
RBCs:
- Hemolytic anemia d/t poor RBC defense against oxidizing agents (eg, fava beans, sulfonamides, primaquine, anti-TB drugs) - Infection can also precipitate hemolysis (free radicals generated by inflammatory response can diffuse into RBCs and cause oxidative damage) |
|
|
What are the implications of a glucose-6P dehydrogenase deficiency?
|
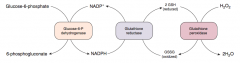
- Glucose 6-P Dehydrogenase is responsible for regenerating NADPH from NADP+
- NADPH is necessary to reduce GSSG (oxidized glutathione) to GSH (reduced glutathione) - GSH is necessary to detoxify free radicals and peroxides |
|
|
What causes glucose-6P dehydrogenase deficiency? Who is more likely to get this?
|
- X-linked recessive disorder
- Most common human enzyme deficiency - More prevalent among blacks as it confers ↑ malarial resistance |
|
|
What does glucose-6P dehydrogenase deficiency cause?
|
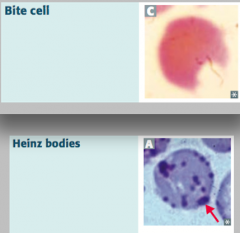
Heinz bodies:
- Oxidized hemoglobin precipitated within RBCs Bite cells: - Results from phagocytic removal of Heinz bodies by splenic macrophages *Think, "Bite into some Heinz ketchup"* |
|
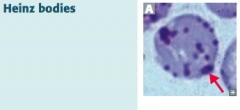
What are Heinz bodies? What are they a sign of?
|
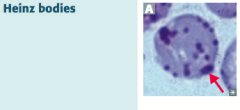
- Oxidized Hemoglobin precipitated within RBCs
- Caused by Glucose-6P Dehydrogenase (G6PD) deficiency (X-linked recessive) |
|

What are bite cells? What are they a sign of?
|
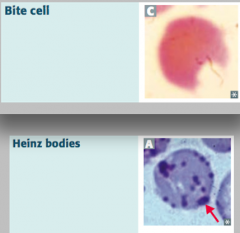
- Result from phagocytic removal of Heinz bodies by splenic macrophages
- Caused by Glucose-6P Dehydrogenase (G6PD) deficiency (X-linked recessive) |
|
|
What are the disorders of fructose metabolism?
|
- Essential fructosuria
- Fructose intolerance |
|
|
What happens in Essential Fructosuria? Cause?
|
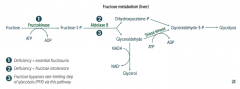
- Defect in Fructokinase (1)
- Autsomal Recessive - Benign, asymptomatic condition, since fructose is not trapped in cells - Fructose appears in blood and urine - Mild condition compared to analogous disorders in galactose metabolism |
|
|
What happens in Fructose Intolerance? Cause? Treatment?
|
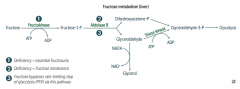
- Hereditary deficiency of Aldolase B (2)
- Autosomal recessive - Fructose-1P accumulates, causing ↓ in available phosophate, which inhibits glycogenolysis and gluconeogenesis - Symptoms occur after consuming fruit, juice, or honey - Urine dipstick will be negative (tests for glucose only); reducing sugar can be detected in urine (nonspecific test for inborn errors of CHO metabolism) - Symptoms: hypoglycemia, jaundice, cirrhosis, vomiting - Tx: ↓ intake of both fructose and sucrose (glucose + fructose) |
|
|
What is the difference in causes of Essential Fructosuria and Fructose Intolerance?
|
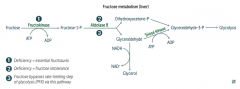
- Essential Fructosuria: defect in Fructokinase, autosomal recessive
- Fructose Intolerance: deficiency of Aldolase B, autosomal recessive; fructose-1P accumulates, causing ↓ availability of phosphate, which inhibits glycogenolysis and gluconeogenesis (less glucose available) |
|
|
What is the difference in symptoms of Essential Fructosuria and Fructose Intolerance?
|
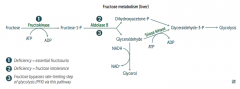
Essential Fructosuria:
- Asymptomatic (fructose is not trapped in cells) - Fructose appears in urine and blood Fructose Intolerance: - Hypoglycemia (d/t inhibition of glycogenolysis and gluconeogenesis) - Jaundice and cirrhosis - Vomiting |
|
|
What are the disorders of Galactose Metabolism?
|

- Galactokinase deficiency
- Classic galactosemia |
|
|
What happens in Galactokinase Deficiency? Cause?
|
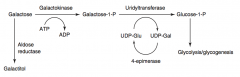
- Hereditary deficiency of Galactokinase
- Autosomal recessive - Galacitol accumulates if galactose present in diet - Relatively mild condition - Galactose appears in blood and urine - Infantile cataracts - May initially present as failure to track objects or to develop a social smile |
|
|
What happens in Classic Galactosemia? Cause? Treat?
|
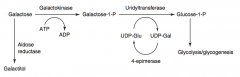
- Absence of Galactose-1-Phosphate Uridyltransferase
- Autosomal Recessive - Damage is caused by accumulation of toxic substances (including galactitol, which accumulates in lens of eye) - Symptoms: failure to thrive, jaundice, hepatomegaly, infantile cataracts, intellectual disability - May lead to E. coli sepsis in neonates - Treatment: exclude galactose and lactose (galactose + glucose) from diet |
|
|
What is the difference in causes of Galactokinase Deficiency and Classic Galactosemia?
|
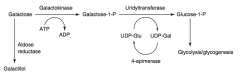
- Galactokinase deficiency: deficiency of galactokinase, galactitol accumulates if galactose present in diet; autosomal recessive
- Classic galactosemia: absence of galactose-1P uridyltransferase, damage is caused by accumulation of toxic substances (galactitol - in lens of eye); depletion of PO4-; autosomal recessive |
|
|
What is the difference in symptoms of Galactokinase Deficiency and Classic Galactosemia?
|
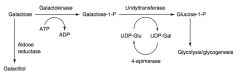
Galactokinase deficiency:
- Mild condition - Galactose appears in blood and urine - Infantile cataracts - may initially present as failure to track objects or to develop a social smile Classic galactosemia: - Failure to thrive - Jaundice - Hepatomegaly - Infantile cataracts (accumulation of galactitol in lens) - Intellectual disability - Can lead to E. coli sepsis in neonates |
|
|
Which of the disorders of fructose deficiency is similar to one of the disorders of galactose deficiency?
|
Fructose Intolerance (d/t deficiency of Aldolase B) is similar to Classic Galactosemia (d/t deficiency of Galactose-1P Uridyltransferase
*FAB-GUT* Fructose is to Aldolase B as Galactose is to Uridyl-Transferase |
|
|
What is an alternative method of trapping glucose in the cell?
|

Convert it to its alcohol counterpart, called Sorbitol, via Aldose Reductase
|
|
|
How is sorbitol synthesized? What can happen to it next?
|

1. Glucose → Sorbitol (via Aldose Reductase and NADPH)
2. Sorbitol → Fructose (via Sorbitol Dehydrogenase and NAD+) 2nd reaction only occurs in certain tissues (liver, ovaries, seminal vesicles) |
|
|
What are the implications if tissues don't have both enzymes to generate sorbitol and to dehydrogenate sorbitol?
|

- Tissues like Schwann cells, retina, the lens, and kidneys only have Aldose Reductase to generate Sorbitol
- These tissues are at risk for intracellular sorbitol accumulation, causing osmotic damage (eg, cataracts, retinopathy, and peripheral neuropathy with chronic hyperglycemia in diabetes) |
|
|
What causes cataracts, retinopathy, and peripheral neuropathy in patients with chronic hyperglycemia / diabetes?
|

- Glucose is converted to Sorbitol via Aldose Reductase
- Schwann cells (→ peripheral neuropathy), retina (→ retinopathy), lens (→ cataracts), and kidneys don't have sufficient Sorbitol Dehydrogenase to remove Sorbitol, leading to OSMOTIC DAMAGE |
|
|
Besides glucose, high levels of what other molecule can also be converted by Aldose Reductase? Product?
|
Galactose → Galactitol (via Aldose Reductase)
|
|
|
What causes Lactose Intolerance?
|
- Insufficient lactase enzyme → dietary lactose intolerance
- Lactase functions on the brush border to digest lactose (in human and cow milk) into glucose and galactose |
|
|
What are the two types of lactase deficiency? How do they differ?
|
Primary:
- Age dependent decline after childhood (Absence of lactase-persistent allele) - Common in Asian, African, or Native American heritage Secondary: - Loss of brush border d/t gastroenteritis (eg, rotavirus), autoimmune disease, etc |
|
|
What are the signs / symptoms of Lactose Intolerance?
|
- Stool demonstrates ↓ pH
- Breath shows ↑ H+ content with lactose tolerance test - Intestinal biopsy reveals normal mucosa in patients with hereditary lactose intolerance - Bloating, cramps, flatulence, osmotic diarrhea |
|
|
How do you treat Lactose Intolerance?
|
Avoid dairy products or add lactase pills to diet
|
|
|
What form of amino acids are found in proteins in humans?
|
Only L-form of amino acids
|
|
|
What type of amino acids need to be supplied in the diet?
|
Essential Amino Acids:
- Glucogenic: Met, Val, His - Glucogenic / Ketogenic: Ile, Phe, Thr, Trp - Ketogenic: Leu, Lys |
|
|
What are the glucogenic essential amino acids?
|
- Methionine (Met)
- Valine (Val) - Histidine (His) |
|
|
What are the glucogenic / ketogenic essential amino acids?
|
- Isoleucine (Ile)
- Phenylalanine (Phe) - Threonine (Thr) - Tryptophan (Trp) |
|
|
What are the ketogenic essential amino acids?
|
- Leucine (Leu)
- Lysine (Lys) |
|
|
What are the acidic amino acids?
|
- Aspartic Acid (Asp)
- Glutamic Acid (Glu) Negatively charged at body pH |
|
|
What are the basic amino acids?
|
- Arginine (Arg) - most basic
- Lysine (Lys) - Histidine (His) - no charge at body pH |
|
|
Which amino acids are required during periods of growth?
|
- Arginine (Arg) - most basic AA
- Histidine (His) - basic, but no charge at body pH |
|
|
Which amino acids are increased in histones? Function?
|
- Arginine (Arg) - most basic AA
- Lysine (Lys) - basic They bind negatively charged DNA |
|
|
What process is necessary for amino acid catabolism?
|
Urea Cycle
|
|
|
What is the function of the Urea Cycle?
|
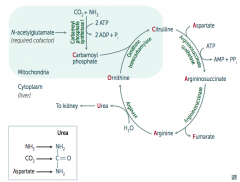
- AA catabolism results in the formation of common metabolites (eg, pyruvates, acetyl-CoA)
- These serve as metabolite fuels - Excess nitrogen (NH3) generated by this process is converted to urea and excreted by the kidneys |
|
|
How do you remember the intermediates in the Urea Cycle?
|
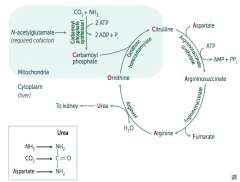
Ordinarily, Careless Crappers Are Also Frivolous About Urination:
- Ornithine + Carbomoyl phosphate → - Citrulline + Aspartate → - Argininosuccinate → - Arginine → (+Fumarate) - Urea→ |
|
|
What are the enzymes in the Urea Cycle?
|
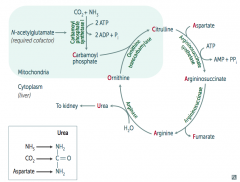
1. Ornithine Transcarbamylase
2. Argininosuccinate Synthetase 3. Argininosuccinase 4. Arginase |
|
|
What is Urea made of?
|
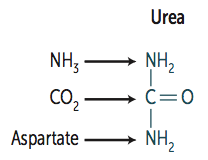
- NH3 (ammonia)
- CO2 - Aspartate (donates NH2) |
|
|
How is Carbamoyl Phosphate synthesized for the Urea Cycle?
|
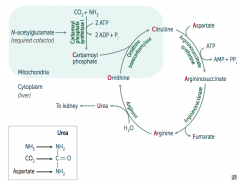
In mitochondria, Carbamoyl Phosphate Synhtetase I combines CO2 + NH3
Uses 2 ATP and requires N-acetylglutamate as a cofactor |
|
|
What happens to Carbamoyl Phosphate in Urea Cycle?
|
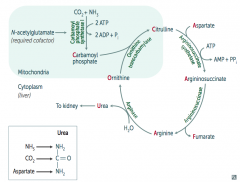
Combines with Ornithine via Ornithine Transcarbamylase → Citrulline
|
|
|
What happens to Citrulline in Urea Cycle?
|
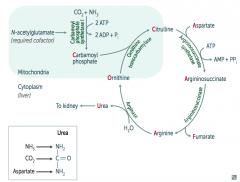
Citrulline combines with Aspartate via Argininosuccinate Synthetase → Argininosuccinate
Requires one ATP → AMP + PPi |
|
|
What happens to Argininosuccinate in Urea Cycle?
|
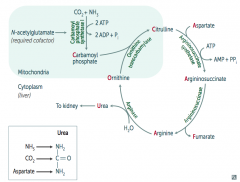
Argininosuccinase breaks it down into Fumarate (released) and Arginine (continues in Urea Cycle)
|
|
|
What happens to Arginine in Urea Cycle?
|
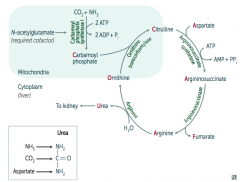
Arginase combines Arginine with H2O to release Urea (which goes to kidney) and Ornithine (regenerated to continue Urea Cycle)
|
|
|
What processes allow safe transport of ammonia in the body?
|
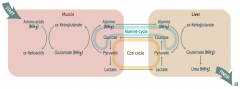
Cori Cycle and Alanine Cycle
|
|
|
How do amino acids (ammonia / NH3) get safely transported from the muscle to the liver (to be converted to urea)?
|
Muscle:
- Amino Acids (NH3) + α-Ketoglutarate → Glutamate (NH3) + α-Ketoacids - Glutamate (NH3) + Pyruvate → Alanine (NH3) + α-Ketoglutarate Alanine Cycle: - Alanine (NH3) transported to liver Liver: - Alanine (NH3) + α-Ketoglutarate → Glutamate (NH3) + Pyruvate - Glutamate (NH3) →→ Urea (NH3) → kidney |
|
|
What is the Alanine Cycle?
|
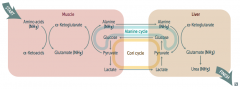
1. Alanine (NH3) transported from muscle to liver
2. Alanine (NH3) + α-Ketoglutarate → Glutamate (NH3) + Pyruvate 3. Pyruvate → Glucose 4. Glucose transported from liver back to muscle 5. Glucose → Pyruvate 6. Pyruvate + Glutamate (NH3) → Alanine (NH3) + α-Ketoglutarate |
|
|
What is the Cori Cycle?
|
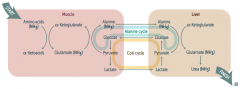
In muscle:
- Glucose → Pyruvate - Pyruvate → Lactate Cori Cycle: - Lactate transported from muscle to liver In liver: - Lactate → Pyruvate - Pyruvate → Glucose (overlaps with Alanine Cycle) Cori / Alanine Cycle: - Glucose transported from liver back to muscle |
|
|
What can cause Hyperammonemia?
|
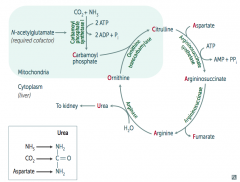
Acquired (eg, liver disease)
Hereditary (eg, urea cycle enzyme deficiency) - N-acetylglutamate deficiency - Ornithine transcarbamylase deficiency - Etc. |
|
|
What does Hyperammonemia cause? Symptoms?
|
- Excess NH4+ → depletes α-Ketoglutarate, leading to inhibition of TCA cycle
- Symptoms: tremor (asterixis), slurring of speech, somnolence, vomiting, cerebral edema, blurring of vision |
|
|
How do you treat Hyperammonemia?
|
- Limit protein in diet
- Benzoate or phenylbutyrate (both of which bind AA and lead to excretion) can be given to ↓ ammonia levels) - Lactulose can acidify the GI tract and trap NH4+ for excretion |
|
|
What are the implications of an N-acetylglutamate deficiency?
|
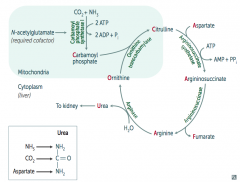
- Required cofactor for Carbamoyl Phosphate Synthetase I
- Absence → Hyperammonemia (because Urea Cycle requires Carbamoyl Phosphate) |
|
|
What is the presentation of an N-acetylglutamate deficiency?
|
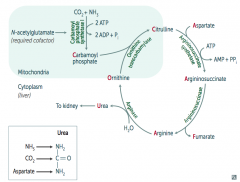
Identical to Carbamoyl Phosphate Synthetase I deficiency:
- Hyperammonemia: tremor (asterixis), slurring of speech, somnolence, vomiting, cerebral edema, blurring of vision - ↑ Ornithine with normal urea cycle enzymes suggests hereditary N-acetylglutamate deficiency |
|
|
What is the most common urea cycle disorder? Cause?
|
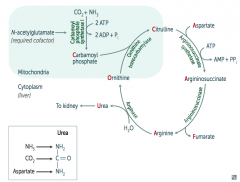
- Ornithine Transcarbamylase Deficiency (which is supposed to combine Carbamoyl Phosphate and Ornithine to make Citrulline)
- X-linked recessive |
|
|
What are the implications of an Ornithine Transcarbamylase Deficiency?
|
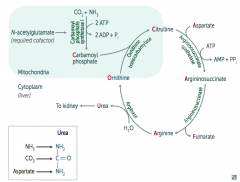
- Interferes with the body's ability to eliminate ammonia
- Often evident in the first few days of life, but may present with late onset - Excess carbamoyl phosphate is converted to orotic acid (part of the pyrimidine synthesis pathway) |
|
|
What are the lab findings of Ornithine Transcarbamylase Deficiency?
|
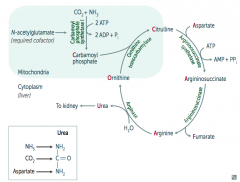
- ↑ Orotic acid in blood and urine (made from excess unused Carbamoyl Phosphate)
- ↓ BUN - Hyperammonemia - No megaloblastic anemia (vs orotic aciduria) |
|
|
What are the amino acid derivatives of Phenylalanine?
|
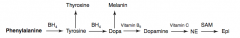
- Tyrosine → Thyroxine
- Melanin - Dopamine → Norepinephrine → Epinephrine |
|
|
What are the amino acid derivatives of Tryptophan?
|
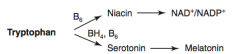
- NAD+ / NADP+
- Serotonin → Melatonin |
|
|
What are the amino acid derivatives of Histidine?
|
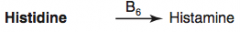
Histamine
|
|
|
What are the amino acid derivatives of Glycine?
|

Porphyrin → Heme
|
|
|
What are the amino acid derivatives of Glutamate?
|
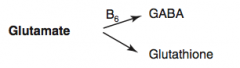
- GABA
- Glutathione |
|
|
What are the amino acid derivatives of Arginine?
|
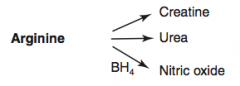
- Creatine
- Urea - Nitric Oxide |
|
|
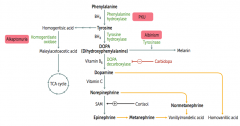
How is Thyroxine (T4) synthesized?
|
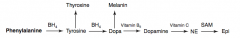
Phenyalanine (BH4) → Tyrosine → Thyroxine
|
|
|
How is Melanin synthesized?
|
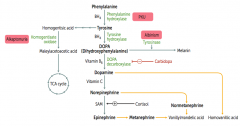
Phenyalanine (BH4) → Tyrosine (BH4) → Dopa → Melanin
Phe → Tyr via Phenylalanine Hydroxylase Tyr → DOPA via Tyrosine Hydroxylase DOPA → Melanin via Tyrosinase |
|
|
How is Dopamine synthesized?
|
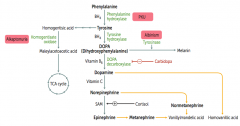
Phenyalanine (BH4) → Tyrosine (BH4) → Dopa (Vitamin B6) → Dopamine
Phe → Tyr via Phenylalanine Hydroxylase Tyr → DOPA via Tyrosine Hydroxylase DOPA → Dopamine via DOPA Decarboxylase |
|
|
How is Norepinephrine synthesized?
|
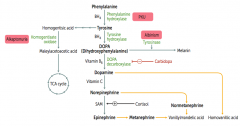
Phenyalanine (BH4) → Tyrosine (BH4) → Dopa (Vitamin B6) → Dopamine (Vitamin C) → Norepinephrine
Phe → Tyr via Phenylalanine Hydroxylase Tyr → DOPA via Tyrosine Hydroxylase DOPA → Dopamine via DOPA Decarboxylase Dopamine → NE |
|
|
How is Epinephrine synthesized?
|
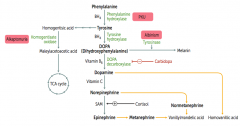
Phenyalanine (BH4) → Tyrosine (BH4) → Dopa (Vitamin B6) → Dopamine (Vitamin C) → Norepinephrine (SAM) → Epinephrine
Phe → Tyr via Phenylalanine Hydroxylase Tyr → DOPA via Tyrosine Hydroxylase DOPA → Dopamine via DOPA Decarboxylase Dopamine → NE requires Vitamin C NE → Epinephrine requires SAM |
|
|
How are NAD+ / NADP+ synthesized?
|
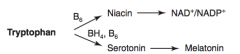
Tryptophan (B6) → Niacin → NAD+ / NADP+
|
|
|
How is Serotonin synthesized?
|
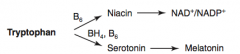
Tryptophan (BH4, B6) → Serotonin
|
|
|
How is Melatonin synthesized?
|
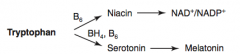
Tryptophan (BH4, B6) → Serotonin → Melatonin
|
|
|
How is Histamine synthesized?
|
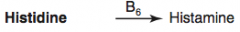
Histidine (B6) → Histamine
|
|
|
How is Heme synthesized?
|

Glycine (B6) → Porphyrin → Heme
|
|
|
How is GABA synthesized?
|
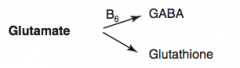
Glutamate (B6) → GABA
|
|
|
How is Glutathione synthesized?
|
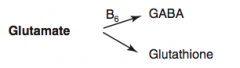
Glutamate → Glutathione
|
|
|
How is Creatinine synthesized?
|
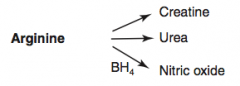
Arginine → Creatinine
|
|
|
How is Urea synthesized?
|
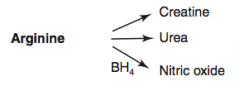
Arginine → Urea
|
|
|
How is Nitric Oxide synthesized?
|
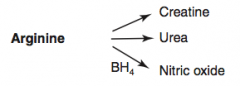
Arginine (BH4) → Nitric Oxide
|
|
|
Which disorder is caused by a deficiency of Phenylalanine Hydroxylase or ↓ BH4 (tetrahydrobiopterin cofactor)?
|
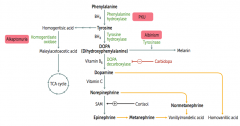
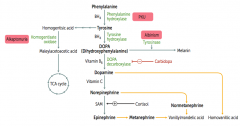
Phenylketonuria
|
|
|
What is the cause of Phenylketonuria?
|
Autosomal recessive:
- Deficiency of Phyenylalanine Hydroxylase OR - Decreased BH4 (tetrahydrobiopterin) cofactor = Malignant PKU * Tyrosine becomes essential |
|
|
What are the symptoms of Phenylketonuria?
|
- Increased phenylalanine leads to excess phenylketones (phenylacetate, phenyllactate, and phenylpyruvate) in urine
- Intellectual disability - Growth retardation - Seizures - Fair skin - Eczema - Musty body odor (disorder or "AROMATIC" AA metabolism) |
|
|
How do you diagnose Phenylketonuria?
|
Screened for 2-3 days after birth (normal at birth because of maternal enzyme during fetal life)
|
|
|
How do you treat Phenylketonuria?
|
- Decrease phenylalanine and increase tyrosine in diet
- Avoid artificial sweetener aspartame because it contains lots of phenyalanine |
|
|
What are the characteristics of Maternal Phenylketonuria (PKU)?
|
- Lack of proper dietary therapy during pregnancy, if mom has PKU, the high levels of phenylalanine can buildup and affect fetus
- Findings in infant: microcephaly, intellectual disability, growth retardation, congenital heart defects |
|
|
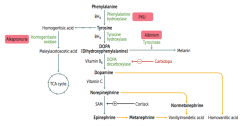
Which disorder is caused by a deficiency of Homogentisate Oxidase?
|
Alkaptonuria (Ochronosis)
|
|
|
What is the cause of Alkaptonuria?
|
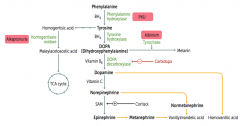
Congenital deficiency of homogentisate oxidase in degradative pathway of tyrosine to fumarate
- Autosomal recessive - Benign disease |
|
|
What are the findings of Alkaptonuria?
|
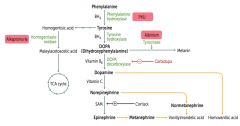
- Dark CT
- Brown pigmented sclerae - Urine turns black on prolonged exposure to air - May have debilitating arthralgias (homogentisic acid is toxic to cartilage) |
|
|
What disease causes intellectual disability, growth retardation, seizures, fair skin, eczema, and MUSTY BODY ODOR?
|
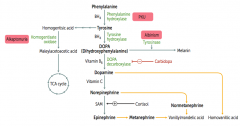
Phenylketonuria - caused by autosomal recessive deficiency of phenylalanine hydroxylase or decreased BH4 cofactor
|
|
|
What disease causes dark CT, brown pigmented sclerae, and urine that turns black on prolonged exposure to air?
|
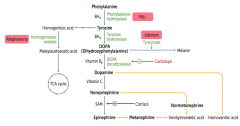
Alkaptonuria - caused by congenital (autosomal recessive) deficiency of Homogentisate Oxidase
|
|
|
What can cause Homocystinuria?
|
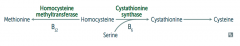
- Cystathionine synthase deficiency
- Decreased affinity of cystathionine synthase for pyridoxal phosphate (PLP) - Homocysteine methyltransferase deficiency |
|
|
What are the implications of cystathionine synthase deficiency, decreased affinity of cystathionine synthease for PLP, or homocysteine methyltransferase deficiency?
|
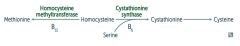
Homocystinuria:
- All forms result in excess homocysteine - ↑↑ homocysteine in urine - Intellectual disability - Osteoporosis - Tall stature - Kyphosis (curvature of spine) - Lens subluxation (downward and inward) - Thrombosis - Atherosclerosis (stroke and MI) |
|
|
Which disorder is caused by a deficiency of tyrosinase?
|
Albinism (prevents conversion of DOPA to Melanin)
|
|
|
What disorder is caused by a defect of the renal PCT and intestinal AA transporter for Cysteine, Ornithine, Lysine, and Arginine (COLA)?
|
Cystinuria
Cystine is made of 2 cysteines connected by a disulfide bond |
|
|
What is the cause of Cystinuria?
|
- Hereditary (autosomal recessive) defect of the renal PCT and intestinal AA transporter for Cysteine, Ornithine, Lysine, and Arginine (COLA)
- Excess cystine in urine can lead to precipitation of hexagonal cystine stones - 1:7000 births |
|
|
How do you diagnose Cystinuria? Treat?
|
- Diagnose: urinary cyanide-nitroprusside test diagnostic
Treatment: - Urinary alkalinization (eg, potassium citrate, acetazolamide) - Chelating agents which increase solubility of cystine stones - Good hydration |
|
|
What disorder is caused by blocked degradation of branched AAs (Isoleucine, Leucine, Valine)? Enzymatic cause?
|
Maple Syrup Urine Disease
- ↓ α-Ketoacid Dehydrogenase (B1) - Causes ↑ α-Ketoacids in the blood, especially those of Leucine - Autosomal recessive |
|
|
What is the cause of Maple Syrup Urine Disease? Implications?
|
- Blocked degradation of branched AAs (Ile, Leu, Val) d/t ↓ α-Ketoacid Dehydrogenase (B1)
- Causes ↑ α-Ketoacids in the blood, especially those of Leucine Symptoms: - Urine smells like maple syrup / burnt sugar - Severe CNS defects - Intellectual disability - Death |
|
|
Which amino acids are not degraded in Maple Syrup Urine Disease?
|
Branched AAs:
- Isoleucine - Leucine - Valine I Love Vermont Maple Syrup from maple trees (with branches) |
|
|
How do you treat Maple Syrup Urine Disease?
|
- Restriction of Isoleucine, Leucine, and Valine in diet
- Thiamine supplementation |
|
|
How does glucagon affect glycogen regulation?
|
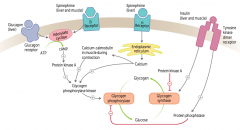
1. Glucagon binds glucagon receptors in liver
2. Activates adenylate cyclase 3. ATP → cAMP 4. cAMP stimulates PKA 5a. Activates Glycogen Phosphorylase Kinase 6a. Activates Glycogen Phosphorylase 7a. Glycogen broken down into glucose 5b. PKA also inhibits Glycogen Synthase 6b. Glucose does not form glycogen |
|
|
How does epinephrine acting on β-receptors affect glycogen regulation?
|
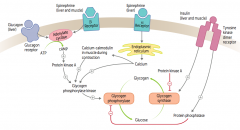
1. Epinephrine binds β-receptors in liver and muscle
2. Activates adenylate cyclase 3. ATP → cAMP 4. cAMP stimulates PKA 5a. Activates Glycogen Phosphorylase Kinase 6a. Activates Glycogen Phosphorylase 7a. Glycogen broken down into glucose 5b. PKA also inhibits Glycogen Synthase 6b. Glucose does not form glycogen |
|
|
How does epinephrine acting on α-receptors affect glycogen regulation?
|
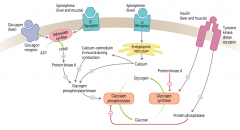
1. Epinephrine binds α-receptors in liver
2. Calcium released from ER 3. Directly and via calcium-calmodulin in muscle during contraction activates Glycogen Phosphorylase Kinase 4. Activates Glycogen Phosphorylase 5. Glycogen broken down into glucose |
|
|
How does insulin affect glycogen regulation?
|
1. Insulin binds Tyrosine Kinase Dimer Receptor in liver and muscle
2a. Activates Glycogen Synthase 3a. Glucose formed into glycogen 2b. Activates Protein Phosphatase 3b. Inhibits Glycogen Phosphorylase 4b. Inhibits breakdown of glycogen into glucose |
|
|
What kind of bonds are in glycogen?
|
- Branches have α-(1,6) bonds
- Linkages have α-(1,4) bonds |
|
|
What happens to glycogen in skeletal muscle?
|
Glycogenolysis → glucose-1P → glucose-6P
Rapidly metabolized during exercise |
|
|
What happens to glycogen in hepatocytes?
|
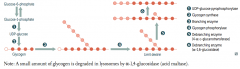
- Glycogen is stored and undergoes glycogenolysis to maintain blood sugar at appropriate levels
- Glycogen phosphorylase cleaves glucose-1P residues off branched glycogen until four remain before a branch point - Then 4α-D-glucanotransferase (debranching enzyme) moves 3 x glucose-1P's from the branch to the linkage - α-1,6-glucosidase (debranching enzyme) cleaves off last glucose-1P on branch |
|
|
How is glycogen synthesized?
|
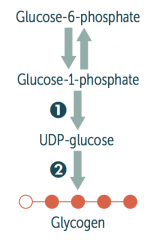
Glucose → Glucose-6P → Glucose-1P
Glucose-1P → UDP-Glucose via UDP-Glucose Pyrophosphorylaes UDP-Glucose → Glycogen via Glycogen Synthase Glycogen can add branches via Branching Enzyme |
|
|
How is glycogen metabolized?
|
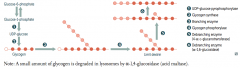
4. Glycogen → Limit Dextrin via Glycogen Phosphorylase (by removing glucose-1P residues until 4 glucose residues on branch remain)
5. 4-α-D-glucanotransferase (debranching enzyme) moves 3 glucose-1Ps from branch to linkage 6. α-1,6-glucosidase (debranching enzyme) cleaves off the last glucose-1P on the branch - Then Glycogen Phosphorylase can continue removing glucose-1P residues |
|
|
A small amount of glycogen is degraded in lysosomes by what enzyme?
|
α-1,4-glucosidase (acid maltase)
|
|
|
What are the glycogen storage diseases? General characteristics?
|
*Very Poor Carbohydrate Metabolism*
- Von Gierke disease (type 1) - Pompe disease (type 2) - Cori disease (type 3) - McArdle disease (type 5) All result in abnormal glycogen metabolism and an accumulation of glycogen within cells; all due to autosomal recessive deficiencies of enzymes |
|
|
What is the cause of Von Gierke disease (type 1 glycogen storage disease)? Findings? Treatment?
|
Cause:
- Glucose-6-Phosphatase deficiency (autosomal recessive) Findings: - Severe fasting hypoglycemia - ↑↑ glycogen in liver - ↑ blood lactate - Hepatomegaly Treatment: - Frequent oral glucose / cornstarch - Avoidance of fructose and galactose |
|
|
What is the cause of Pompe disease (type 2 glycogen storage disease)? Findings? Other?
|
Cause:
- Lysosomal α-1,4-glucosidase (acid maltase) - Autosomal recessive Findings: - Cardiomyopathy - Systemic findings leading to early death Comments: - Pompe trashes the Pump (heart, liver, and muscles) |
|
|
What is the cause of Cori disease (type 3 glycogen storage disease)? Findings? Other?
|
Cause:
- Debranching enzyme (α-1,6-glucosidase) deficiency - Autosomal recessive Findings: - Milder form of type 1 (Von Gierke) - Normal blood lactate levels Comments: - Gluconeogenesis is intact |
|
|
What is the cause of McArdle disease (type V glycogen storage disease)? Findings? Other?
|
Cause:
- Skeletal muscle glycogen phosphorylase (myophosphorylase) deficiency - Autosomal recessive Findings: - ↑ glycogen in muscle, but can't break it down - Leads to painful muscle cramps, myoglobinuria (red urine) w/ strenuous exercise, and arrhythmia from electrolyte abnormalities Comments: - McArdle = Muscle |
|
|
Which disease presents with severe fasting hypoglycemia, ↑↑ glycogen in liver, ↑ blood lactate, and hepatomegaly? Cause?
|
Von Gierke Disease (type 1 Glycogen Storage Disease)
- Glucose-6-Phosphatase Deficiency - Autosomal recessive - Treatment: frequent oral glucose/cornstarch, avoid fructose and galactose |
|
|
Which disease presents with cardiomyopathy and systemic findings that lead to an early death? Cause?
|
Pompe Disease (type 2 glycogen storage disease)
- Lysosomal α-1,4-glucosidase (acid maltase) deficiency - Autosomal recessive - Pompe trashes the Pump (heart, liver, and muscle) |
|
|
Which disease presents as a milder form of Von Gierke Disease with normal blood lactate levels? Cause?
|
Cori Disease (type 3 glycogen storage disease)
- Debranching enzyme (α-1,6-glucosidase) deficiency - Autosomal recessive - Gluconeogenesis is still intact |
|
|
Which disease presents with painful muscle cramps, myoglobinuria (red urine) w/ strenuous exercise, and arrhythmia? Cause?
|
McArdle Disease (type 5 glycogen storage disease)
- Skeletal muscle glycogen phosphorylase (myophosphorylase) deficiency - Autosomal recessive - ↑ Glycogen in muscle that can't be broken down causes the severe muscle cramps - Arrhythmia caused by electrolyte abnormalities - McArdle = Muscle |
|
|
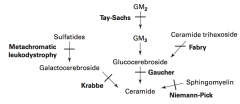
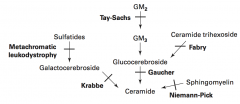
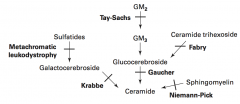
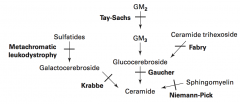
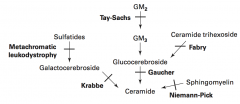
What are the lysosomal storage diseases? General characteristics?
|
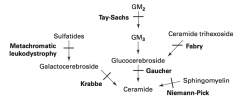
Sphingolipidoses:
- Fabry Disease - Gaucher Disease - Niemann-Pick Disease - Tay-Sachs Disease - Krabbe Disease - Metachromatic Leukodystrophy Mucopolysaccharidoses: - Hurler Syndrome - Hunter Syndrome |
|
|
What is the cause of Fabry disease (lysosomal storage disease)? Findings? What accumulates?
|
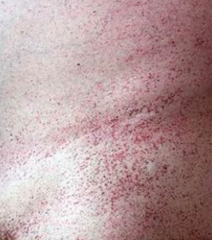
Cause:
- α-Galactosidase A deficiency - X-linked recessive Findings: - Peripheral neuropathy of hands/feet - Angiokeratomas (benign cutaneous lesion of capillaries, resulting in small marks of red to blue color) - CV / Renal disease Accumulation: - Ceramide Trihexoside |
|
|
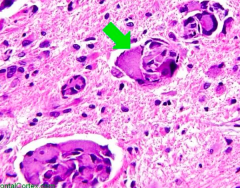
What is the cause of Gaucher disease (lysosomal storage disease)? Findings? What accumulates? Treatment?
|
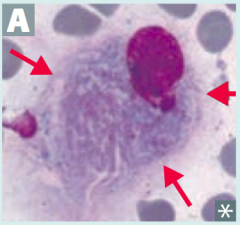
Cause:
- Glucocerebrosidase (β-glucosidase) deficiency - Autosomal recessive Findings: - Most common - Hepatosplenomegaly - Pancytopenia - Aseptic necrosis of femur and bone crises - Gaucher cells (picture) - lipid laden macrophages resembling crumpled tissue paper Accumulation: - Glucocerebroside Treatment: - Recombinant Glucocerebrosidase |
|
|
What is the cause of Niemann-Pick disease (lysosomal storage disease)? Findings? What accumulates?
|
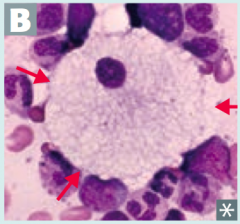
Cause:
- Sphingomyelinase deficiency - Autosomal Recessive Findings: - Progressive neurodegeneration - Hepatosplenomegaly - "Cherry red" spot on macula - Foam cells (lipid-laden macrophages) - picture Accumulation: - Sphingomyelin *No Man Picks (Niemann Pick) his nose with his SPHINGer (sphingomyelinase deficiency)* |
|
|
What is the cause of Tay-Sachs disease (lysosomal storage disease)? Findings? What accumulates?
|
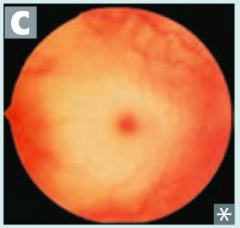
Cause:
- Hexosaminidase A deficiency - Autosomal Recessive Findings: - Progressive neurodegeneration - Developmental delay - "Cherry red" spot on macula - Lysosomes with onion skin - No Hepatosplenomegaly (vs Niemann Pick) Accumulation: - GM2 Ganglioside *Tay-SaX lacks heXosaminidase* |
|
|
What is the cause of Krabbe disease (lysosomal storage disease)? Findings? What accumulates?
|
Cause:
- Galactocerebrosidase deficiency - Autosomal recessive Findings: - Peripheral neuropathy - Developmental delay - Optic atrophy - Globoid cells - picture Accumulation: - Galactocerebroside - Psychosine |
|
|
What is the cause of Metachromatic Leukodystrophy (lysosomal storage disease)? Findings? What accumulates?
|
Cause: |
|
|
What is the cause of Hurler Syndrome (lysosomal storage disease)? Findings? What accumulates?
|
Cause:
- α-L-iduronidase deficiency - Autosomal Recessive Findings: - Developmental delay - Gargoylism - Airway obstruction - Corneal clouding - Hepatosplenomegaly Accumulation: - Heparan sulfate - Dermatan sulfate |
|
|
What is the cause of Hunter Syndrome (lysosomal storage disease)? Findings? What accumulates?
|
Cause:
- Iduronate Sulfatase deficiency - X-linked Recessive Findings: - Mild Hurler Syndrome + aggressive behavior - No corneal clouding Accumulation: - Heparan sulfate - Dermatan sulfate *Hunters see clearly (no corneal clouding) and aggressively aim for the X (X-linked recessive* |
|
|
Which lysosomal storage disorders are increased in Ashkenazi Jews?
|
- Tay-Sachs
- Niemann-Pick - Some forms of Gaucher disease |
|
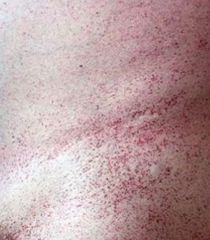
Which disease presents with peripheral neuropathy of hands/feet, angiokeratomas (picture), and CV/renal disease? Cause?
|
Fabry Disease
- Deficiency of α-Galactosidase A - Accumulate Ceramide Trihexoside - X-linked recessive |
|
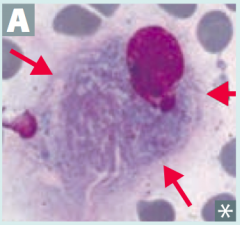
What is the most common lysosomal storage disorder, causing hepatosplenomegaly, pancytopenia, aseptic necrosis of femur, bone crises, and lipid-laden macrophages (picture)? Cause?
|
Gaucher Disease
- Deficiency of Glucocerebrosidase (β-glucosidase) - Accumulate Glucocerbroside - Autosomal recessive |
|
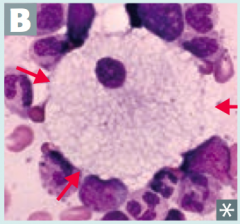
Which disease presents with progressive neurodgeneration, hepatosplenomegaly, cherry red spot on macula, and foam cells (lipid-laden macrophages (picture)? Cause?
|
Niemann-Pick Disease
- Deficiency of Sphingomyelinase (*No Man Picks (Niemann Pick) his nose with his SPHINGer*) - Accumulation of sphingomyelin - Autosomal recessive |
|
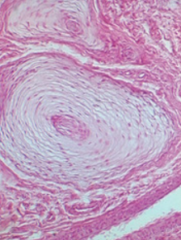
Which disease presents with progressive neurodegeneration, developmental delay, cherry red spot on macula, lysosomes with onion skin (picture), and NO hepatosplenomegaly? Cause?
|
Tay-Sachs Disease
- Deficiency of Hexosaminidase A (*Tay-SaX lacks heXosaminidase*) - Accumulation of GM2 ganglioside - Autosomal recessive |
|
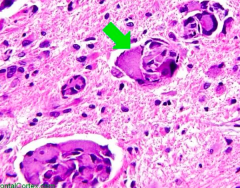
Which disease presents with peripheral neuropathy, developmental delay, optic atrophy, and globoid cell (picture)? Cause?
|
Krabbe Disease
- Deficiency of Galactocerebrosidase - Accumulation of Galactocerebroside and Psychosine - Autosomal Recessive |
|
|
Which disease presents with central and peripheral demyelination with ataxia and dementia? Cause?
|
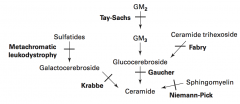
Metachromatic Leukodystrophy
- Deficiency of Arylsulfatase A - Accumulation of Cerebroside Sulfate - Autosomal recessive |
|
|
Which disease presents with developmental delay, gargoylism, airway obstruction, corneal clouding, and hepatosplenomegaly? Cause?
|
Hurler Syndrome
- Deficiency of α-L-iduronidase - Accumulation of heparan sulfate and dermatan sulfate - Autosomal recessive |
|
|
Which disease presents as a mild form of Hurler syndrome with aggressive behavior, but no corneal clouding? Cause?
|
Hunter Syndrome
- Deficiency of iduronate sulfatase - Accumulation of heparan sulfate and dermatan sulfate - X-linked recessive *Hunters see clearly (no corneal clouding) and aggressively aim for the X (X-linked recessive* |
|
|
What is required for fatty acid metabolism / degradation?
|
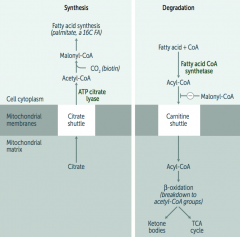
Carnitine-dependent transport into the mitochondrial matrix
|
|
|
What is Carnitine used for? What happens if it is deficient / symptoms?
|
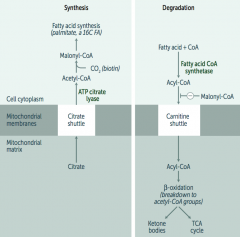
- Carnitine is necessary for long-chain FA degradation
(CARnitine = CARnage of FAs) - Deficiency: inability to transport LCFAs into the mitos, resulting in toxic accumulation; causes weakness, hypotonia, and hypoketotic hypoglycemia |
|
|
What transport shuttle is required for fatty acid synthesis?
|
Citrate Shuttle ("SYtrate" = SYnthesis)
|
|
|
What happens if there is an Acyl-CoA Dehydrogenase deficiency?
|
- ↑ Dicarboxylic Acids
- ↓ Glucose and Ketones - Acetyl-CoA is a (+) allosteric regulator of pyruvate carboxylase in gluconeogenesis - ↓ Acetyl-CoA → ↓ fasting glucose |
|
|
What causes formation of Ketone Bodies?
|
- In prolonged starvation and diabetic ketoacidosis, oxaloacetate is depleted for gluconeogenesis
- In alcoholism, excess NADH shunts oxaloacetate to malate * Both processes cause a build-up of Acetyl-CoA, which shunts glucose and FFA toward production of ketone bodies |
|
|
What are the ketone bodies? What are they made from?
|
Ketone Bodies: Acetoacetate + β-Hydroxybutyrate
- To be used in muscle and brain - Made in liver from fatty acids and amino acids |
|
|
What are the signs and symptoms of Ketone Body accumulation?
|
- Breath smells like acetone (fruity color)
- Urine test for ketones does not detect β-hydroxybutyrate |
|
|
What should you think if the patient's breath smells like acetone?
|
Check their urine for ketone bodies
|
|
|
How many calories are in 1g of protein, carbohydrate, fat, and alcohol?
|
- 1g protein = 4 kcal
- 1g carbohydrate = 4 kcal - 1g alcohol = 7 kcal - 1g fat = 9 kcal |
|
|
What are the sources of fuel for exercise? How long do they last? How does overall performance change with different fuel uses?
|
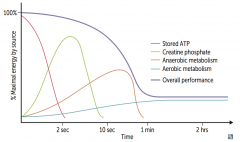
Maximum performance
- <2 seconds: Stored ATP - 2-10 seconds: Creatinine Phosphate Moderate performance - 10 sec - 1 min: Anaerobic Metabolism Low performance - >1 minute: Aerobic Metabolism |
|
|
During fasting and starvation, what are the priorities of the body?
|
- Supply sufficient glucose to the brain and RBCs (RBCs lack mitochondria and so cannot use ketones)
- Preserve protein |
|
|
What happens to metabolism during the fed state (after a meal)?
|
- Glycolysis and aerobic respiration
- Insulin stimulates storage of lipids, proteins, and glycogen |
|
|
What happens to metabolism during the fasting state (between meals)?
|
- Hepatic glycogenolysis (major)
- Hepatic gluconeogenesis and adipose release of FFAs (minor) - Glucagon and adrenaline stimulate use of fuel reserves |
|
|
What happens to metabolism after 1-3 days of starvation?
|
Blood glucose levels maintained by:
- Hepatic glycogenolysis - Adipose release of FFA - Muscle and liver shift fuel use from glucose to FFA - Hepatic gluconeogenesis from peripheral tissue lactate and alanine, and from adipose tissue glycerol and prionyl-CoA (from odd-chain FFA - the only TAG components that contribute to gluconeogenesis) Glycogen reserves are depleted after 1 day RBCs lack mitochondria and so cannot use ketones |
|
|
What happens in the liver after 1-3 days of starvation?
|
- Hepatic glycogenolysis
- Shifts fuel use from glucose to FFAs - Hepatic gluconeogenesis from peripheral tissue lactate and alanine, and from adipose tissue glycerol and propionyl-CoA (from odd-chain FFA) |
|
|
What happens in the adipose after 1-3 days of starvation?
|
- Release of FFA
- FFA used in liver for gluconeogenesis |
|
|
What happens in the muscle after 1-3 days of starvation?
|
- Shifts fuel use from glucose to FFA
- Lactate and alanine are sent to liver for gluconeogenesis |
|
|
What happens to metabolism after 3 days of starvation?
|
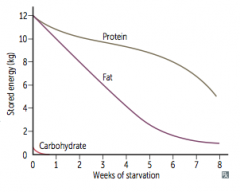
- Adipose stores form ketone bodies - main source of energy for brain
- After depletion of adipose stores, vital protein degradation accelerates, leading to organ failure and death - Amount of excess stores determines survival time |
|
|
What is the rate-limiting step in cholesterol synthesis? Regulation?
|
HMG-CoA Reductase: converts HMG-CoA to Mevalonate
- Induced by insulin - Inhibited by Statin drugs (eg, Lovastatin) competitively and reversibly |
|
|
What happens to plasma cholesterol?
|
2/3 is esterified by lecithin-cholesterol acyltranferase (LCAT)
|
|
|
What enzymes degrade lipids?
|
- Pancreatic Lipase: degrades dietary TGs in small intestine
- Lipoprotein Lipase (LPL): degrades TGs circulating in chylomicrons and VLDLs (found on vascular endothelial surface) - Hepatic TG Lipase (HL): degrades TG remaining in IDL - Hormon-Sensitive Lipase: degrades TG stored in adipocytes |
|
|
Which enzyme degrades dietary TGs in small intestine?
|
Pancreatic Lipase
|
|
|
Which enzyme degrades TG circulating in chylomicrons and VLDLs? Where is it found?
|
Lipoprotein Lipase (LPL) - found on vascular endothelial surface
|
|
|
Which enzyme degrades TG remaining in IDL?
|
Hepatic TG Lipase (HG)
|
|
|
Which enzyme degrades TG stored in adipocytes?
|
Hormone-Sensitive Lipase
|
|
|
What is the action of LCAT (Lecithin-Cholesterol Acyl-Transferase)?
|
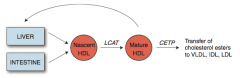
Catalyzes esterification of cholesterol
Converts nascent HDL to mature HDL |
|
|
What is the action of CETP?
|
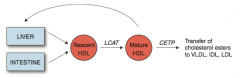
Cholesterol Ester Transfer Protein
- Mediates transfer of cholesterol esters to other lipoprotein particles |
|
|
What are the functions of lipoproteins?
|
- Lipoproteins are composed of varying proportions of cholesterol, TGs, and phospholipids
- LDL and HDL carry most cholesterol - LDL transports cholesterol from liver to tissues (LDL = Lousy) - HDL transports cholesterol from periphery to liver (HDL = Healthy) |
|
|
What is the function of Chylmicrons? Source?
|
- Delivers dietary TGs to peripheral tissue
- Delivers cholesterol to liver in the form of chylomicron remnants, which are mostly depleted for the TAGs - Secreted by intestinal epithelial cells |
|
|
What is the function of VLDL? Source?
|
- Delivers hepatic TGs to peripheral tissue
- Secreted by liver |
|
|
What is the function of IDL? Source?
|
- Formed in the degradation of VLDL
- Delivers TGs and cholesterol to liver |
|
|
What is the function of LDL? Source?
|
- Delivers hepatic cholesterol to peripheral tissues
- Formed by hepatic lipase modification of IDL in the peripheral tissue - Taken up by target cells via receptor-mediated endocytosis |
|
|
What is the function of HDL? Source?
|
- Mediates reverse cholesterol transport from periphery to liver
- Acts as a repository for apoC and apoE (which are needed for chylomicron and VLDL metabolism) - Secreted from both liver and intestine - Alcohol increases synthesis |
|
|
What are the types of familial dyslipidemias?
|
- I: hyperchylmicronemia
- IIa: familial hypercholesterolemia - IV: hyper-triglyceridemia |
|
|
What happens in type I familial dyslipidemia? Cause?
|
Hyperchylmicronemia
- Increased in blood: chylomicrons, TG, and cholesterol - Pancreatitis - Hepatosplenomegaly - Eruptive / prurithic xanthomas (no increased risk for atherosclerosis) Cause: autosomal recessive deficiency of lipoprotein lipase or altered apolipoprotein C-II |
|
|
What happens in type IIa familial dyslipidemia? Cause?
|
Familial Hypercholesterolemia
- Increased in blood: LDL and cholesterol - Heterozygotes have cholesterol ~ 300 mg/dL - Homozygotes have cholesterol ~ 700+ mg/dL - Causes accelerated atherosclerosis (may have MI before age 20) - Tendon (Achilles) xanthomas - Corneal arcus Cause: autosomal dominant absent or defective LDL receptors |
|
|
What happens in type IV familial dyslipidemia? Cause?
|
Hyper-Triglyceridemia
- Elevated in blood: VLDL and TG - Pancreatitis Cause: autosomal dominant, hepatic overproduction of VLDL |
|
|
Which disease is caused by lipoprotein lipase deficiency or an altered apolipoprotein C-II?
|
Hyperchylmicronemia / Type I familial dyslipidemia:
- Increased in blood: chylomicrons, TG, and cholesterol - Pancreatitis - Hepatosplenomegaly - Eruptive / prurithic xanthomas (no increased risk for atherosclerosis) (Autosomal Recessive) |
|
|
Which disease is caused by absent or defective LDL receptors?
|
Familial Hypercholesterolemia / type IIa familial dyslipidemia:
- Increased in blood: LDL and cholesterol - Heterozygotes have cholesterol ~ 300 mg/dL - Homozygotes have cholesterol ~ 700+ mg/dL - Causes accelerated atherosclerosis (may have MI before age 20) - Tendon (Achilles) xanthomas - Corneal arcus (Autosomal Dominant) |
|
|
Which disease is caused by hepatic overproduction of VLDL?
|
Hyper-Triglyceridemia / type IV Familial Dyslipidemia
- Elevated in blood: VLDL and TG - Pancreatitis (Autosomal Dominant) |
|
|
Which disease is characterized by pancreatitis, hepatosplenomegaly, and eruptive pruritic xanthoams, with no increased risk for atherosclerosis? Cause?
|
Hyperchylmicronemia
- Increased in blood: chylomicrons, TG, and cholesterol - Pancreatitis - Hepatosplenomegaly - Eruptive / prurithic xanthomas (no increased risk for atherosclerosis) Cause: autosomal recessive deficiency of lipoprotein lipase or altered apolipoprotein C-II |
|
|
Which disease is characterized by high cholesterol (~300 mg/dL if heterozygous and ~700+ if homozygous) with accelerated atherosclerosis and possibly MI before age 20; tendon (Achilles) xanthomas and corneal arcus?
|
Familial Hypercholesterolemia
- Increased in blood: LDL and cholesterol - Heterozygotes have cholesterol ~ 300 mg/dL - Homozygotes have cholesterol ~ 700+ mg/dL - Causes accelerated atherosclerosis (may have MI before age 20) - Tendon (Achilles) xanthomas - Corneal arcus Cause: autosomal dominant absent or defective LDL receptors |
|
|
Which disease is characterized by hepatic overproduction of VLDL and causes pancreatitis?
|
Hyper-Triglyceridemia
- Elevated in blood: VLDL and TG - Pancreatitis Cause: autosomal dominant, hepatic overproduction of VLDL |