Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
51 Cards in this Set
- Front
- Back
|
How are NAD+ / NADP+ synthesized?
|
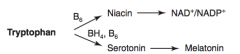
Tryptophan (B6) → Niacin → NAD+ / NADP+
|
|
|
How is Serotonin synthesized?
|
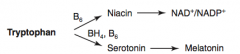
Tryptophan (BH4, B6) → Serotonin
|
|
|
How is Melatonin synthesized?
|
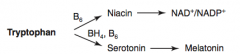
Tryptophan (BH4, B6) → Serotonin → Melatonin
|
|
|
How is Histamine synthesized?
|
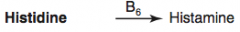
Histidine (B6) → Histamine
|
|
|
How is Heme synthesized?
|

Glycine (B6) → Porphyrin → Heme
|
|
|
How is GABA synthesized?
|
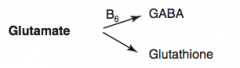
Glutamate (B6) → GABA
|
|
|
How is Glutathione synthesized?
|
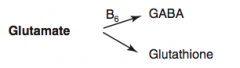
Glutamate → Glutathione
|
|
|
How is Creatinine synthesized?
|
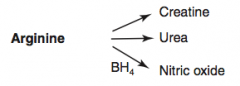
Arginine → Creatinine
|
|
|
How is Urea synthesized?
|
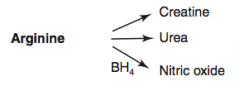
Arginine → Urea
|
|
|
How is Nitric Oxide synthesized?
|
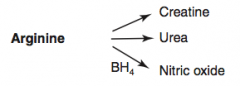
Arginine (BH4) → Nitric Oxide
|
|
|
Which disorder is caused by a deficiency of Phenylalanine Hydroxylase or ↓ BH4 (tetrahydrobiopterin cofactor)?
|
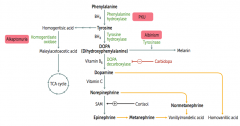
Phenylketonuria
|
|
|
What is the cause of Phenylketonuria?
|
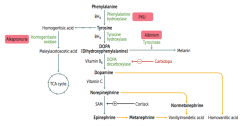
Autosomal recessive:
- Deficiency of Phyenylalanine Hydroxylase OR - Decreased BH4 (tetrahydrobiopterin) cofactor = Malignant PKU * Tyrosine becomes essential |
|
|
What are the symptoms of Phenylketonuria?
|
- Increased phenylalanine leads to excess phenylketones (phenylacetate, phenyllactate, and phenylpyruvate) in urine
- Intellectual disability - Growth retardation - Seizures - Fair skin - Eczema - Musty body odor (disorder or "AROMATIC" AA metabolism) |
|
|
How do you diagnose Phenylketonuria?
|
Screened for 2-3 days after birth (normal at birth because of maternal enzyme during fetal life)
|
|
|
How do you treat Phenylketonuria?
|
- Decrease phenylalanine and increase tyrosine in diet
- Avoid artificial sweetener aspartame because it contains lots of phenyalanine |
|
|
What are the characteristics of Maternal Phenylketonuria (PKU)?
|
- Lack of proper dietary therapy during pregnancy, if mom has PKU, the high levels of phenylalanine can buildup and affect fetus
- Findings in infant: microcephaly, intellectual disability, growth retardation, congenital heart defects |
|
|
Which disorder is caused by a deficiency of Homogentisate Oxidase?
|
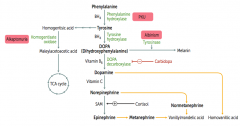
Alkaptonuria (Ochronosis)
|
|
|
What is the cause of Alkaptonuria?
|
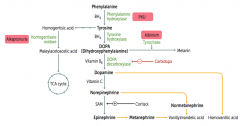
Congenital deficiency of homogentisate oxidase in degradative pathway of tyrosine to fumarate
- Autosomal recessive - Benign disease |
|
|
What are the findings of Alkaptonuria?
|
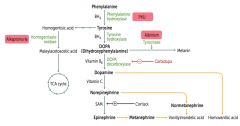
- Dark CT
- Brown pigmented sclerae - Urine turns black on prolonged exposure to air - May have debilitating arthralgias (homogentisic acid is toxic to cartilage) |
|
|
What disease causes intellectual disability, growth retardation, seizures, fair skin, eczema, and MUSTY BODY ODOR?
|
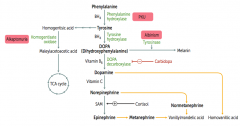
Phenylketonuria - caused by autosomal recessive deficiency of phenylalanine hydroxylase or decreased BH4 cofactor
|
|
|
What disease causes dark CT, brown pigmented sclerae, and urine that turns black on prolonged exposure to air?
|
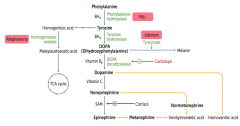
Alkaptonuria - caused by congenital (autosomal recessive) deficiency of Homogentisate Oxidase
|
|
|
What can cause Homocystinuria?
|
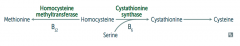
- Cystathionine synthase deficiency
- Decreased affinity of cystathionine synthase for pyridoxal phosphate (PLP) - Homocysteine methyltransferase deficiency |
|
|
What are the implications of cystathionine synthase deficiency, decreased affinity of cystathionine synthease for PLP, or homocysteine methyltransferase deficiency?
|
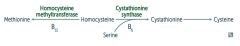
Homocystinuria:
- All forms result in excess homocysteine - ↑↑ homocysteine in urine - Intellectual disability - Osteoporosis - Tall stature - Kyphosis (curvature of spine) - Lens subluxation (downward and inward) - Thrombosis - Atherosclerosis (stroke and MI) |
|
|
Which disorder is caused by a deficiency of tyrosinase?
|
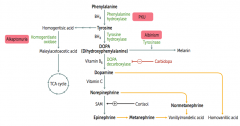
Albinism (prevents conversion of DOPA to Melanin)
|
|
|
What disorder is caused by a defect of the renal PCT and intestinal AA transporter for Cysteine, Ornithine, Lysine, and Arginine (COLA)?
|
Cystinuria
Cystine is made of 2 cysteines connected by a disulfide bond |
|
|
What is the cause of Cystinuria?
|
- Hereditary (autosomal recessive) defect of the renal PCT and intestinal AA transporter for Cysteine, Ornithine, Lysine, and Arginine (COLA)
- Excess cystine in urine can lead to precipitation of hexagonal cystine stones - 1:7000 births |
|
|
How do you diagnose Cystinuria? Treat?
|
- Diagnose: urinary cyanide-nitroprusside test diagnostic
Treatment: - Urinary alkalinization (eg, potassium citrate, acetazolamide) - Chelating agents which increase solubility of cystine stones - Good hydration |
|
|
What disorder is caused by blocked degradation of branched AAs (Isoleucine, Leucine, Valine)? Enzymatic cause?
|
Maple Syrup Urine Disease
- ↓ α-Ketoacid Dehydrogenase (B1) - Causes ↑ α-Ketoacids in the blood, especially those of Leucine - Autosomal recessive |
|
|
What is the cause of Maple Syrup Urine Disease? Implications?
|
- Blocked degradation of branched AAs (Ile, Leu, Val) d/t ↓ α-Ketoacid Dehydrogenase (B1)
- Causes ↑ α-Ketoacids in the blood, especially those of Leucine Symptoms: - Urine smells like maple syrup / burnt sugar - Severe CNS defects - Intellectual disability - Death |
|
|
Which amino acids are not degraded in Maple Syrup Urine Disease?
|
Branched AAs:
- Isoleucine - Leucine - Valine I Love Vermont Maple Syrup from maple trees (with branches) |
|
|
How do you treat Maple Syrup Urine Disease?
|
- Restriction of Isoleucine, Leucine, and Valine in diet
- Thiamine supplementation |
|
|
How does glucagon affect glycogen regulation?
|
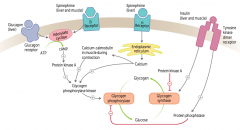
1. Glucagon binds glucagon receptors in liver
2. Activates adenylate cyclase 3. ATP → cAMP 4. cAMP stimulates PKA 5a. Activates Glycogen Phosphorylase Kinase 6a. Activates Glycogen Phosphorylase 7a. Glycogen broken down into glucose 5b. PKA also inhibits Glycogen Synthase 6b. Glucose does not form glycogen |
|
|
How does epinephrine acting on β-receptors affect glycogen regulation?
|
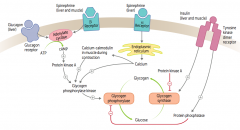
1. Epinephrine binds β-receptors in liver and muscle
2. Activates adenylate cyclase 3. ATP → cAMP 4. cAMP stimulates PKA 5a. Activates Glycogen Phosphorylase Kinase 6a. Activates Glycogen Phosphorylase 7a. Glycogen broken down into glucose 5b. PKA also inhibits Glycogen Synthase 6b. Glucose does not form glycogen |
|
|
How does epinephrine acting on α-receptors affect glycogen regulation?
|
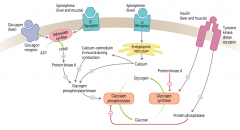
1. Epinephrine binds α-receptors in liver
2. Calcium released from ER 3. Directly and via calcium-calmodulin in muscle during contraction activates Glycogen Phosphorylase Kinase 4. Activates Glycogen Phosphorylase 5. Glycogen broken down into glucose |
|
|
How does insulin affect glycogen regulation?
|
1. Insulin binds Tyrosine Kinase Dimer Receptor in liver and muscle
2a. Activates Glycogen Synthase 3a. Glucose formed into glycogen 2b. Activates Protein Phosphatase 3b. Inhibits Glycogen Phosphorylase 4b. Inhibits breakdown of glycogen into glucose |
|
|
What kind of bonds are in glycogen?
|
- Branches have α-(1,6) bonds
- Linkages have α-(1,4) bonds |
|
|
What happens to glycogen in skeletal muscle?
|
Glycogenolysis → glucose-1P → glucose-6P
Rapidly metabolized during exercise |
|
|
What happens to glycogen in hepatocytes?
|
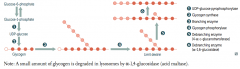
- Glycogen is stored and undergoes glycogenolysis to maintain blood sugar at appropriate levels
- Glycogen phosphorylase cleaves glucose-1P residues off branched glycogen until four remain before a branch point - Then 4α-D-glucanotransferase (debranching enzyme) moves 3 x glucose-1P's from the branch to the linkage - α-1,6-glucosidase (debranching enzyme) cleaves off last glucose-1P on branch |
|
|
How is glycogen synthesized?
|
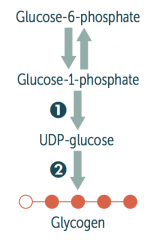
Glucose → Glucose-6P → Glucose-1P
Glucose-1P → UDP-Glucose via UDP-Glucose Pyrophosphorylaes UDP-Glucose → Glycogen via Glycogen Synthase Glycogen can add branches via Branching Enzyme |
|
|
How is glycogen metabolized?
|
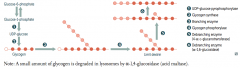
4. Glycogen → Limit Dextrin via Glycogen Phosphorylase (by removing glucose-1P residues until 4 glucose residues on branch remain)
5. 4-α-D-glucanotransferase (debranching enzyme) moves 3 glucose-1Ps from branch to linkage 6. α-1,6-glucosidase (debranching enzyme) cleaves off the last glucose-1P on the branch - Then Glycogen Phosphorylase can continue removing glucose-1P residues |
|
|
A small amount of glycogen is degraded in lysosomes by what enzyme?
|
α-1,4-glucosidase (acid maltase)
|
|
|
What are the glycogen storage diseases? General characteristics?
|
*Very Poor Carbohydrate Metabolism*
- Von Gierke disease (type 1) - Pompe disease (type 2) - Cori disease (type 3) - McArdle disease (type 5) All result in abnormal glycogen metabolism and an accumulation of glycogen within cells; all due to autosomal recessive deficiencies of enzymes |
|
|
What is the cause of Von Gierke disease (type 1 glycogen storage disease)? Findings? Treatment?
|
Cause:
- Glucose-6-Phosphatase deficiency (autosomal recessive) Findings: - Severe fasting hypoglycemia - ↑↑ glycogen in liver - ↑ blood lactate - Hepatomegaly Treatment: - Frequent oral glucose / cornstarch - Avoidance of fructose and galactose |
|
|
What is the cause of Pompe disease (type 2 glycogen storage disease)? Findings? Other?
|
Cause:
- Lysosomal α-1,4-glucosidase (acid maltase) - Autosomal recessive Findings: - Cardiomyopathy - Systemic findings leading to early death Comments: - Pompe trashes the Pump (heart, liver, and muscles) |
|
|
What is the cause of Cori disease (type 3 glycogen storage disease)? Findings? Other?
|
Cause:
- Debranching enzyme (α-1,6-glucosidase) deficiency - Autosomal recessive Findings: - Milder form of type 1 (Von Gierke) - Normal blood lactate levels Comments: - Gluconeogenesis is intact |
|
|
What is the cause of McArdle disease (type V glycogen storage disease)? Findings? Other?
|
Cause:
- Skeletal muscle glycogen phosphorylase (myophosphorylase) deficiency - Autosomal recessive Findings: - ↑ glycogen in muscle, but can't break it down - Leads to painful muscle cramps, myoglobinuria (red urine) w/ strenuous exercise, and arrhythmia from electrolyte abnormalities Comments: - McArdle = Muscle |
|
|
Which disease presents with severe fasting hypoglycemia, ↑↑ glycogen in liver, ↑ blood lactate, and hepatomegaly? Cause?
|
Von Gierke Disease (type 1 Glycogen Storage Disease)
- Glucose-6-Phosphatase Deficiency - Autosomal recessive - Treatment: frequent oral glucose/cornstarch, avoid fructose and galactose |
|
|
Which disease presents with cardiomyopathy and systemic findings that lead to an early death? Cause?
|
Pompe Disease (type 2 glycogen storage disease)
- Lysosomal α-1,4-glucosidase (acid maltase) deficiency - Autosomal recessive - Pompe trashes the Pump (heart, liver, and muscle) |
|
|
Which disease presents as a milder form of Von Gierke Disease with normal blood lactate levels? Cause?
|
Cori Disease (type 3 glycogen storage disease)
- Debranching enzyme (α-1,6-glucosidase) deficiency - Autosomal recessive - Gluconeogenesis is still intact |
|
|
Which disease presents with painful muscle cramps, myoglobinuria (red urine) w/ strenuous exercise, and arrhythmia? Cause?
|
McArdle Disease (type 5 glycogen storage disease)
- Skeletal muscle glycogen phosphorylase (myophosphorylase) deficiency - Autosomal recessive - ↑ Glycogen in muscle that can't be broken down causes the severe muscle cramps - Arrhythmia caused by electrolyte abnormalities - McArdle = Muscle |
|
|
What are the lysosomal storage diseases? General characteristics?
|
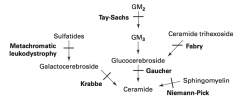
Sphingolipidoses:
- Fabry Disease - Gaucher Disease - Niemann-Pick Disease - Tay-Sachs Disease - Krabbe Disease - Metachromatic Leukodystrophy Mucopolysaccharidoses: - Hurler Syndrome - Hunter Syndrome |