Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
578 Cards in this Set
- Front
- Back
- 3rd side (hint)
|
What are the most common forms of endocrine disease?
|
1. Diabetes Mellitus
2. Diabetes Insipidus 3. Thyroid disease 4. Acromegaly 5. Cushing's disease |
|
|
|
What is the incidence of Diabetes Mellitus?
|
50-150/1000
|
|
|
|
What is the incidence of Thyroid disease?
|
5-120/1000
|
|
|
|
What is the incidence of prolactinoma?
|
0.140/1000
|
|
|
|
What is the incidence of MEN1?
|
0.03-0.2/1000
|
|
|
|
What is the incidence of acromegaly?
|
0.003/1000
|
|
|
|
What is the incidence of Cushing's disease?
|
0.002-0.005/1000
|
|
|
|
What is the incidence of severe CAH?
|
0.067/1000
|
|
|
|
What is the incidence of mild CAH?
|
1/1000
|
|
|
|
What is CAH?
|
>Congenital adrenal hyperplasia
|
|
|
|
How do hormones act on cells to generate an effect?
|
>Either through action at the plasma membrane through second messengers
- thereby having an effect on second messengers >Or by diffusion through the membrane, binding to their receptor and interacting with a hormone response element on the gene of interest to have their effect |
|
|
|
What are examples of hormones binding and activating nuclear receptors?
|
>Lipophilic substances such as:
- endogenous hormones - vitamins A and D - xenobiotic endocrine disruptors >Because the expression of a large number of genes is regulated by nuclear receptors, ligands that activate these receptors can have profound effects on the organism - many of these regulated genes are associated with various diseases, which explains why the molecular targets of approx. 13% of USFDA approved drugs are nuclear |
|
|
|
What are examples of lipid soluble hormones?
|
1. Glucocorticoids
- increase blood sugar - antiinflammatory action (cortisol) 2. Mineralocorticoids - maintain water and salt balance e.g. aldosterone 3. Oestrogens - involved in female sex development (β-oestradiol) 4. Androgens - required for male sex development (testosterone) 5. Vitamin D - required for bone development and calcium metabolism (D3) 6. Thyroid hormones - controls basal metabolic rate (T3) 7. Retinoic acids - Morphogen |
|
|
|
What are nuclear receptors, and how do they function?
|
>A class of proteins found within cells that are responsible for sensing steroid and thyroid hormones and certain other molecules
- they work with other proteins to regulate the expression of specific genes - thus controlling the development, homeostasis and metabolism of the organism >Hydrophobic hormones that are lipid soluble and therefore can pass the cell membrane interact with superfamily of receptors that are intracellular (cytosolic or nuclear) - receptors thereby function as hormone regulated transcription factors, controlling the expression of specific target genes by interacting with HREs, located close to the promoter in the regulatory region |
|
|
|
What differentiates nuclear receptors from other classes of receptor?
|
>They have the ability to directly bind to DNA and regulate the expression of adjacent genes
- hence these receptors are classified as transcription factors >More specifically, ligand binding to a nuclear receptor results in a conformational change in the receptor - in turn activating the receptor, resulting in up or downregulation of gene expression >They are able to directly interact with and control the expression of genomic DNA - as a consequence, nuclear receptors play key roles in both embryonic development and adult homeostasis |
|
|
|
What is the mechanism for transcription activation by nuclear receptors?
|
>Through increased recruitment of pol 11
- causes chromatin modification |
|
|
|
What are the broad classes of nuclear receptor?
|
>Types I, II and III
|
|
|
|
What ligands activate type I nuclear receptors?
|
LIPOPHILIC
1. Glucocorticoid (receptor) - cortisol, hydrocortisol - increase blood sugar, antiinflammatory 2. Progesterone (receptor) - progesterone - sex hormone 3. Oestrogen (receptor) - oestrogen - sex hormone 4. Androgen (receptor) - testosterone, dihydrotestosterone - sex hormone |
|
|
|
How are type I nuclear receptors activated?
|
>Ligand binding to type I nuclear receptors in the cytosol results in the dissociation of heat shock proteins, homo-dimerisation, translocation (i.e. active transport) from the cytoplasm into the cell nucleus, and binding to specific sequences of DNA known as HREs
>Type I nuclear receptors bind to HREs consisting of two half sites separated by a variable length of DNA, and the second half site has a sequence inverted from the first (inverted repeat) - type I nuclear receptors include members of subfamily 3 such as the androgen, oestrogen, glucocorticoid and progesterone receptors |
|
|
|
What is the structure of nuclear receptors?
|
>Nuclear receptors are modular in structure and contain the following domains:
N TERMINAL REGULATORY DOMAIN: - contains the activation function 1 (AF-1), whose action is independent of the presence of ligand - the transcriptional activation of AF-1 is normally very weak, but it does synergise with AF-2 in the E domain, to produce a more robust upregulation of gene expression A-B DOMAIN: - highly variable in sequence between various nuclear receptors DNA BINDING DOMAIN: - Highly conserved domain containing two zinc fingers that binds to specific sequences of DNA called HREs HINGE REGION: - Thought to be a flexible domain that connects the DBD with the ligand binding domain - influences intracellular trafficking and subcellular distribution LIGAND BINDING DOMAIN: - moderately conserved in sequence and highly conserved in structure between the various nuclear receptors - the structure of the LBD is referred to as an alpha helices (the sandwich filling) - along with the DBD, the LBD contributes to the dimerisation interface of the receptor and in addition binds coactivator and corepressor proteins >The LBD also constainst the activation function 2, whose action is dependent on the presence of bound ligand C-TERMINAL DOMAIN: - Highly variable in sequence between various nuclear receptors |
|
|
|
What are Type II nuclear receptors? How do thesy differ from type I?
|
>In contrast to type I, type II receptors are retained in the nucleus regardless of the ligand binding status
- and in addition bind as hetero-dimers (usually with RXR) to DNA - in the absence of ligand, type II nuclear receptors are often complexed with corepressor proteins - ligand binding to the nuclear receptor causes dissociation of corepressor and recruitment of coactivator proteins - additional proteins including RNA polymerase are then recruited to the NR/DNA complex that transcribes DNA into messenger RNA >Type II receptors in contrast to type I, bind to direct repeat instead of inverted repeats |
|
|
|
What are examples of type II receptors?
|
1. Thyroid hormone receptors - T3
2. Vitamin D3 - 1,25 dihydroxyvitamin D3 3. Retinoic acid - 9 cisretinoic acid 4. All transretinoic acid |
|
|
|
What are the characteristics of type III receptors?
|
>Orphan receptors
- the endogenous ligand is not yet known - similar to type one, bind to DNA as homodimers - however bind to direct repeats instead of inverted repeats >Some of these receptors, such as FXR, LXR and PPAR bind a number of metabolic intermediates such as fatty acids, bile acids and or sterols with relatively low affinity - these receptors hence may function as metabolic sensors >Other nuclear receptors such as CAR and PXR appear to function as xenobiotic sensors upregulating the expression of CYP450 enzymes that metabolise these xenobiotics |
|
|
|
What is a zinc finger?
|
>Zinc finger (Znf) domains are relatively small protein motifs which contain multiple finger-like protrusions that make tandem contacts with their target molecule.
- Some of these domains bind zinc, but many do not, instead binding other metals such as iron, or no metal at all |
|
|
|
What is a direct repeat?
|
>Direct repeats are a type of genetic sequence that consists of two or more repeats of a specific sequence
|
|
|
|
What is an inverted repeat?
|
>An inverted repeat (or IR) is a sequence of nucleotides that is the reversed complement of another sequence further downstream
What are type IV nuclear receptors? >Bind as monomers, or dimers but only a singler DNA binding domain of the receptor binds to single half site |
|
|
|
What are the transcriptional activities of nuclear receptors?
|
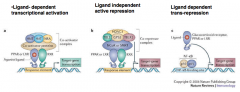
Ligand dependent transcriptional activation
>Ligand independent active repression >Ligand dependent transrepression |
LIGAND DEPENDENT TRANSCRIPTION
|
|
|
What are examples of endocrine related diseases?
|
>Breast cancer
>Prostate cancer >Ovarian cancer >Diabetes >Obesity |
|
|
|
What occurs if there are ER, PR, AR knockouts?
|
>Multiple reproductive abnormalities
|
|
|
|
What occurs if there are RXR knockouts?
|
>Embryonic and post embryonic development
|
|
|
|
What occurs if there are PPARs, LXR knockouts?
|
>Glucose and lipid metabolism
|
|
|
|
What occurs if there are GR knockouts?
|
>Long term spatial memory, stress erythropoiesis, energy metabolism
|
|
|
|
Hyper proinsulinaemia, hypoparathyroidism, dwarfism, hypothyroidism and obesity are all examples of genetic disorders due to mutation in?
|
>Protein hormones
|
|
|
|
Lipid congenital adrenal hyperplasia, CAH: androgen excess, androgen deficiency hypertension, male pseudohermaphroditism, vitamin D resistant rickets are all examples of endocrine disorders with mutations in?
|
>Steroid synthetic enzymes
|
|
|
|
Complete androgen resistance, thyroid hormone resistance, vitamin D resistant rickets are all examples of endocrine diseases due to mutation in?
|
>Nuclear receptors
|
|
|
|
Insulin resistance, hypogonadism, chondrodysplasia, adrenal insufficiency, obesity are all examples of endocrine dsorders due to mutations in?
|
>Membrane receptor mutations (inactivating)
|
|
|
|
What are the non-genomic actions of steroid hormones?
|
>Rapid
>Do not involve direct bindng of steroid receptors to response elements in DNA >Interaction with classical steroid receptors or G protein coupled receptors localised within the plasma membrane |
|
|
|
What are hydrophilic hormones?
|
>Proteins, e.g. insulin, peptides, or amino acid derivatives e.g. adrenaline
>Dissolve in the plasma without needing transport by binding proteins, although there are exceptions (half catecholamines are loosely bound to plasma albumen) >Act by altering intracellular proteins, because hydrophillic hormones can't cross the plasma membrane, they act by binding to receptors on the outer plasma membrane surface of the target cell >Binding of the hormone to the receptor causes an increase in the concentration of 'second messenger' in the cell, which alters the activity of other proteins |
|
|
|
How are hormones classified?
|
ENDOCRINE:
- Act on cells far from the site of release - secreted into the blood - only target cells express the receptor e.g. insulin and adrenaline PARACRINE: - Act on nearby cells only - diffuse in the interstitial fluid and are rapidly inactivated by local enzymes e.g. histamine JUXTACRINE: - hormone either bound to the membrane (requires physical contact between cells - or hormone is secreted into the extracellular matrix AUTOCRINE: - act on the cell that released the hormone - T cells and IL-2 |
|
|
|
Name four different hormone receptor types.
|
1. Ligand gated ion channels e.g. Ach receptor
- signal is transduced to the cell via the change in membrane potential etc, when the ion channel is opened 2. Receptor enzymes - e.g. insulin receptor - enzymatic activity of receptor is activated by hormone binding 3. Enzyme recruiting receptors - e.g. cytokine receptors - hormone binding induces the recruitment and activation of protein kinases 4. G-protein coupled receptors e.g. adrenaline receptors - hormone binding activates GTP binding hormones |
|
|
|
What are GPCRs?
|
>Largest class of cell surface receptors
- several thousand known - widely used as drug targets >Involved in responses to hormones, neurotransmitters, odours, tastes and light >Hormone bound receptor causes the exchange of GDP for GTP, activating the G alpha subunit - 7 alpha helices - the dissociated G alpha subunit then interacts with an enzyme, until it hydrolyses the GTP to GDP, becoming inactive again (takes seconds to minutes) |
|
|
|
What occurs when a GPCR becomes activated?
|
>The 3/4 and 5/6 cytosolic loops interact with G proteins
>The Galpha subunits can be divided into four groups - Gs activates adenylyl cyclase, increasing [cAMP] - Gi inhibits adenlylyl cyclase, decreasing [cAMP] - Gq activates PLC, increasing [DAG], [IP3] and [Ca++] >There are 5 Gbeta and 6 Ggamma isoforms >Tissues regulate their expression of these subunits >The Gbeta-gamma complexes may alter specificity of receptor G-protein binding, cooperate in transduction, or shut the pathway down |
|
|
|
What is the structure of the beta adrenergic receptor pathway?
|
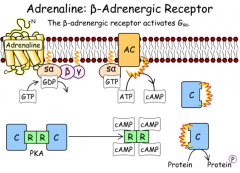
|
B ADRENERGIC RECEPTOR
|
|
|
What effect does pertussis toxin have on G proteins?
|
>Causes AD ribosylation of the Gia subunit, preventing it from interacting with the GPCR, it cannot be activated
|
|
|
|
What is protein kinase A?
|
>A serine / threonine kinase
>Binding of 4x cAMP to the two R subunits causes them to dissociate from the catalytic subunits, activating them >Protein kinase A phosphorylates several enzymes, such as hormone sensitive lipase (+), acetyl CoA carboxylase (-), glycogen synthase (-), and the transcription factor CREB (+) >Protein kinase A is thus able to immediately alter metabolic pathways and have longer term effects via gene transcription |
|
|
|
For example, what are the diverse effects of cAMP - PkA activation on different target proteins?
|
1. Liver (adrenaline, Nad, glucagon)
- increase glycogenolysis and gluconeogenesis 2. Adipose tissue (adrenaline, ACTH) - increase in lipolysis 3. Ovarian follicles (FSH, LH) - increased synthesis of oestrogen and progesterone |
|
|
|
What are the points of amplification in the signal transduction cascade?
|
1. Adrenaline:receptor complex is able to catalyse GDP-GTP exchange on multiple G-proteins
- each activated Galpha subunit cand only bind to one adenylyl cyclase 2. Each active adenylyl cyclase can catalyse the formation of many molecules of cAMP - it takes 4 molecules of cAMP to activate 2xPKA subunits 3. Each active PKA subunit can phosphorylate many proteins |
|
|
|
What are receptor enzymes?
|
>These receptors have an extracellular ligand-binding domain, and an enzyme active site on the intracellular section, connected by a single transmembrane segment
- Many of thse enzymes are tyrosine kinases (RTKs) e.g. the insulin receptor - some have serine / threonine kinase activity - another group have guanylyl cyclase activity (GTP to cGMP) >Binding ligand either activates the enzyme activity or brings it closer to its target |
|
|
|
What are receptor tyrosine kinases>
|
>Function as dimers, with an extracellular hormone-binding domain, and an intracellular protein tyrosine kinase domain
>Upon binding of hormone (many are growth factors), RTK monomers cross-phosphorylate each other >Phosphorylation of the RTK makes it a site of attachment for proteins with SH2 domains, or PTB domains - localising proteins at the membrane >For the insulin receptor, cross phosphorylation causes the kinase to become fully active |
|
|
|
What are epidermal growth factor receptors?
|
1. Binding of EGF to each EGFR monomer induces a structural change that allows the monomers to dimerize
- proximity of the cytosolic domains allows cross phosphorylation 2. Tyrosine phosphates act as docking sites for Grb-2, which is attached to Sos 3. Sos catalysis the exchange of GDP for GTP on membrane-bound Ras, activating it 4. GTP:Ras binds and activates Raf, a membrane bound protein kinase 5. A series of protein kinsases are phosphorylated and activated, resulting in phosphorlation of several transcription factors, altering their activity |
|
|
|
How is EFGR signalling involved in cancer?
|
>EGFR overexpressed in some epithelial cancers
>A small amount of receptor can dimerise in the absence of ligand >Since the tyrosine kinase activity is already present, this is enough to initiate signal transduction, thereby sending an inappropriate 'grow and divide' signal to the cell >A therapeutic antibody (cetuximab) targets the extracellular domain of the receptor, sterically blocking the ability of the receptor to dimerise - successfully used in colorectal cancers |
|
|
|
How does insulin receptor signalling occur?
|
1. Binding of insulin to the dimeric receptor forces the PTK domains together, followed by cross-phosphorylation
2. The first round of cross phosphorylation fully activates the kinase activity and is followed by more cross-phosphorylation 3. These phosphorylated tyrosine residues act as docking sites for IRS-1 (insulin receptor substrate 1), which gets phosphorylated 4. Phosphorylated IRS-1 can bind PI-3K (phosphoinositide-3 kinase) which not located at the membrane, phosphorylates PIP2 at position 3, forming PIP3 5. PIP3 allows both PDK1 and PKB to associate with the membrane via their PH (pleckstrin homology) domains) 6. Phosphorylated PKB dissociates fromt he membrane and phosphorylates its target proteins |
|
|
|
What is IRS-1 and what is important about it?
|
>It is a docking protein as it can bind many proteins, including Grb2 (thereby activating the MAPK pathway)
>There are other proteins that can assemble at the phosphorylated insulin receptor, including IRS2, a homologous protein >Insulin is therefore capable of simultaneously stimulating numerous pathways, involving short term and long term effects |
|
|
|
What is TGF-β?
|
>Transforming growth factor β
>These signalling proteins normally prevent proliferation of most mammalian cells by inducing synthesis of proteins that inhibit the cell cycle >Most mammalian cells secrete at least one TGF-β isoform, and have receptors on their surface >BMP induces bone formation in cultured cells and is now used clinically to strengthen bone fractures >TGFβ proteins also play a role in tissue organisation, promoting expression of extracellular matrix proteins and adhesion molecules |
|
|
|
What is the structure of the TGFβ receptor?
|
>TGFβ binds to TBR-II, whose serine / threonine kinase activity is constitutively active
>This allows it to bind to TBR-I, and phosphorylate its glycine-serine rich GS domeain, activating the S/T kinase activity >TBR-I can then phosphorylate a class of transcription factors called R-Smads >Upon phosphorylation, two R-Smads and a co-Smad form a heterotrimer, and the nuclear localisation signals are also exposed - in the nucleus, the heterotrimer interacts with transcription factors to cause expression of particular target genes |
|
|
|
How is the TGFβ relevant to cancerous cells?
|
>The TGFβ receptor pathway often inhibits growth in cells
- loss of either TBRI or TBRII function due to inactivating mutations is found in many human tumours - these tumours are resistant to growth inhibition by TGFβ >Mutations in the Smad proteins also prevent TGFβ signalling, most human pancreatic cancers contain a deletion in Smad 4 (a co Smad) |
|
|
|
What are cytokine receptors?
|
>Cytokines are a family of small (~160 aas) signalling molecules, with a characteristic arrangement of four alpha helices, controlling the growth and differentiation of a number of cells
>Cytokine receptors do not have an intrinsic enzyme activity, rather they recruit an enzyme >The receptors all have a tyrosine kinase called JAK bound to their cytosolic domains, which phosphorylate transcription members of the Signal Transduction and Activation of Transcription (STAT) family >Although cytokine receptors can activate other pathways e.g. the MAPK pathway, the JAK/STAT pathway is normally only activated by cytokines |
|
|
|
What is an example of a cytokine released in response to low oxygen?
|
>Erythropoeitin - released by the kidney
- stimulates the transcription of genes in erythroid progenitors that prevent them from undergoing apoptosis, and stimulate them to differentiate into erythrocytes - use of supplemental erythropoietin to increase the level of erythrocytes in the blood is banned in international athletic competitions - the use of supplemental erythropoietin is also dangerous as the surplus erythrocytes can clot small blood vessels - several athletes have died of stroke during exercise due to erythropoietin doping |
|
|
|
What is the structure of the erythropoietin receptor?
|
>A JAK2 kinase with low activity is bound to the cytosolic domain of EpoR
>Epo simultaneously binds two EpoRs, bringing the JAK kinases close enough for each to phosphorylate the activation lip of the other - this lowers the Km of the kinase for its substrate activating it >The JAK kinases phosphorylate the receptors allow STAT5 to bind (via SH2 domains), and also get phosphorylated >The phosphorylated STAT5s dissociate from the receptor, dimerise, exposing a nuclear localisation sequence >The STAT5 dimer enters the nuclear nucleus and its DNA binding domain binds to specific DNA regulatory sequences to control the expression of target genes |
|
|
|
What is the JAK/STAT pathway?
|
>SHP1 is a phosphotyrosine phosphatase that binds the phosphorylated receptor and dephosphorylates JAK kinase, inhibiting the pathway when cytokines are no longer binding to the receptor
>A mutant version of the erythropoietin receptor was discovered in an athlete that caused them to have higher levels of RBCs than normal, despite unusually low levels of erythropoietin >This mutant receptor was missing some of the tyrosines normally phosphorylated during signal transduction >The receptor was able to bind and activate STAT5, but was unable to bind the SHP1 phosphatase, resulting in increased intracellular signalling in the erythroid progenitor cells, and more RBCs than usual |
|
|
|
What effect does cholera toxin have on G proteins?
|
>Causes ADP ribosylation of Gsa, resulting in constantly active adenylate cyclase in intestinal epithelial cells
|
|
|
|
What is the structure of the pancreas?
|
>Exocrine - accessory digestive (acinar cells)
- bulk of the pancreas >Endocrine - Islets of Langherhans - small percentage >Lies deep to the stomach, tail close to the spleen, head encircled by the duodenum - 80% lobular - acinar cells - 4% ducts - intercalated ducts to form pancreatic duct - fuses with common bile duct - 2% islet cells >Covered in thin connective tissue capsule which extends inward as septa, partitioning the gland into lobules |
|
|
|
What is the autonomic innervation to the Islets of Langerhans?
|
>Sympathetic adrenergic input
- splanchic nerve from the coeliac plexus >Parasympathetic cholinergic input - vagus nerve |
|
|
|
What 3 cell types are found in the IoL? What do they produce?
|
α cells - glucagon
β cells - insulin δ cells - somatostatin |
|
|
|
What is the structure of insulin?
|
>51 aa polypeptide
- α and β chains joined by disulphide bonds - proinsulin in the ER is exposed to endopeptidases which excise the C peptide, thereby generating the mature form of insulin - packaged in the golgi into secretory granules which accumulate in the cytoplasm - requires Zn and Ca++ |
|
|
|
What is the structure of glucagon?
|
>29 aas long
- produced from preproglucagon, cleaved to active substance - doesn’t require Zn and Ca++, although their presence decrease clearance |
|
|
|
What is the structure of somatostatin?
|
>14aas long
- physiological role unclear - can suppress insulin and glucagon - inhibitory factor - stimulates inhibitory G proteins upon binding GPCRs |
|
|
|
Which factors stimulate insulin secretion?
|
MIXED MEAL
1. Increased plasma [glucose] 2. Increased [free aa] 3. Increased [GI hormones] (gastrin, secretin, CCK, GIP) OTHER FACTORS a. increased [glucagon] b. noradrenaline (low []: α adrenergic receptors) c. acetylcholine |
|
|
|
Which factors inhibit insulin secretion?
|
1. Decreased [glucose]
2. Increased [somatostatin] (pancreatic and gastric) 3. Noradrenaline - (high []: β adrenergic receptors) 4. Adrenaline - (β adrenergic receptors) |
|
|
|
Which factors stimulate glucagon secretion?
|
1. Decreased [glucose]
2. Increased [free aa] 3. Adrenaline |
|
|
|
Which factors inhibit glucagon secretion?
|
An increase [glucose]
|
|
|
|
Which factors stimulate somatostatin secretion?
|
MIXED MEAL
1. Carbohydrates 2. Fats 3. Proteins 4. Decreased pH in duodenum (bulbogastrone mechanism) |
|
|
|
What are the two important effects of insulin?
|
1. Facilitation of entry of glucose into muscle, adipose and several other tissues
2. Stimulates the liver to store glucose in the form of glycogen |
|
|
|
What is the relationship between glycogen synthase/glycogen phosphorylase?
|
>Catalyse a potentially futile cycle, regulated in a reciprocal fashion
- both enzymes can be converted between active and less active forms using a system of protein kinases >Hormones such as glucagon and adrenaline that promote glycogen breakdown act partly via cyclic AMP and PKA - PKA phosphorylates and activates another protein kinase called Phosphorylase B kinase which in turn activates glycogen phosphorylase - resulting in glycogen breakdown to glucose 1 phosphate, and simultaneous phosphorylateion of glycogen synthase, which prevents the futile cycling of g-1-p back into glycogen via UDP glucose |
|
|
|
What is IMGD?
|
>Insulin mediated glucose disposal
- 85% is skeletal and adipose tissue - insulin increases uptake of glucose - glucose --> g6p - G-6-P --> glycogen / TAG |
|
|
|
How does glucose enter cells in the presence of insulin?
|
FACILITATED DIFFUSION
>Hexose transporters e.g. GLUT 4 (major muscle transporter) Absence of insulin - GLUT4 stored in cytoplasmic vesicles Actions of insulin - Fusion of vesicles - Insertion of glucose transporters in the plasma membrane |
|
|
|
What is the structure of a hexose transporter?
|
>Large integral membrane proteins
>Similar structures - 12 membrane spanning regions - cytoplasmic C and N terminal tails - Glycosylated on one of the extracellular loops |
|
|
|
Where are SGLUT 1 receptors found?
|
>GI tract cotransporter with sodium, cotransports glucose or galactose along with 2Na+ (not fructose)
|
|
|
|
Where are GLUT 1 receptors found?
|
>Brain, erythrocyte, endothelial cells, fetal tissues
- glucose and galactose |
|
|
|
Where are GLUT 2 receptors found?
|
>Liver, pancreatic beta cells, small intestines, kidneys
- transports glucose, galactose and fructose - low affinity, high capacity transporter, serves as a glucose sensor in pancreatic beta cells |
|
|
|
Where are GLUT 3 receptors found?
|
>Brain, placenta and testes
- transports glucose (high affinity) and galactose, not fructose - primary glucose transporter for neurons |
|
|
|
Where are GLUT 4 receptors found?
|
>Skeletal and cardiac muscle, adipocytes
THE INSULIN RESPONSIVE GLUCOSE TRANSPORTER - high glucose affinity |
|
|
|
Where are GLUT 5 receptors found?
|
>Small intestine, sperm
- transports fructose, but not glucose or galactose - present also in brain, kidney, adipocytes and muscle |
|
|
|
What stimulates hepatic glycogen storage?
|
>Insulin
- activates hexokinase - phosphorylates glucose, trapping it into the cell >Activates several enzymes directly involved in glycogen synthesis (including phosphofructokinase and glycogen synthase) - inhibits activity of glucose 6 phosphatase >Insulin informs liver to store as much glucose as possible for later use |
|
|
|
What is required enzymatically for glycogen breakdown?
|
>Glycogen synthase and Glycogen phosphorylase
- must be converted between active and less ative forms via protein kinases >PKA phosphorylates and activates PKB, which activates GP >PKA phosphorylates and inactivates GS which prevents cycling of G1P - glycogen breakdown |
|
|
|
What switches on glycogen phosphorylase?
|
>Glucagon activates AC
>Increases cAMP >Activates cAMP dependent kinase (PKA) >Activates phosphorylase kinase >Activates GP (glycogen phosphorylase) - glycogen phosphorylase b (GPb) --> glycogen phosphorylase a |
|
|
|
What switches off glycogen synthetase?
|
>Glucagon switches off glycogen synthetase (GS)
- via cAMP dependent kinase |
|
|
|
What switches on glycogen synthetase?
|
>Insulin
- inhibiting cAMP independent kinase - enhancing phosphoprotein phosphatase |
|
|
|
What is the effect of insulin in the liver?
|
>Promotes glycogenesis
|
|
|
|
What is the effect of glucagon on the liver?
|
>Promotes glycogenolysis
- via glycogen phosphorylase |
|
|
|
What is the effect of insulin on lipid metabolism?
|
>ANABOLIC
- increases net TAG synthesis by - promoting synthesis of fatty acids in the liver - inhibiting breakdown of fat in adipose tissue ACTIVATES - acetyl CoA carboxylase INACTIVATES - hormone sensitive lipase (HSL) |
|
|
|
What is the effect of glucagon on lipid metabolism?
|
>CATABOLIC
- stimulate net breakdown of TAG stores (sparing glucose) INACTIVATES - acetyl CoA carboxylase ACTIVATES - hormone sensitive lipase |
|
|
|
How is glucose and lipid metabolism linked?
|
>Pyruvate enters CAC - citrate can exit for FA biosynthesis (role for acetyl-CoA
>Glycolysis generates alpha phosphoglycerate - backbone of TAG >Pentose phosphate pathway generates reduced cofactors for FA biosynthesis |
|
|
|
Which hormone switches off acetyl CoA carboxylase?
|
>Glucagon, insulin switches it on
|
|
|
|
Which drugs can enhance Hormone sensitive lipase?
|
>Glucocorticoids
- cause redistribution of fats around the body |
|
|
|
What are incretins?
|
>The incretins are hormones that work to increase insulin secretion
- the incretin concept was developed when it was observed that there is substantially more insulin secreted in response to oral glucose versus intravenous - it was hypothesised that glucose in the digestive tract activated a feedforward mechanism that increased insulin secretion, anticipating the rise in blood glucose that would occur following ingestion of carbohydrates DEFINITION: >Naturally occuring hormones that the gut releases throughout the day - the level of active incretins increases significantly when food is ingested >Endogenous incretins GLP-1 and GIP (glucose-dependent insulinotropic peptide) facilitate the response of the pancreas and liver to glucose fluctuations through their actions on pancreatic alpha and beta cells - when glucose levels are elevated, both GLP-1 and GIP signal beta cells to increase insulin release and GLP-1 signals alpha cells to suppress glucagon release - physiologic activity of incretins is limited by the enzyme dipeptidyl peptidase 4 (DPP4) which rapidly degrades active incretins after their release |
|
|
|
How may GLP-1 be interfered with pharmacologically?
|
>Exenatide (Byetta) is a GLP-1 receptor agonist approved for adjunctive therapy for patients with DM 2 who are not well controlled on oral agents
- it is available only as an injection (twice daily) >DPP-4 inhibitors or gliptins - block dipeptidyl peptidase-4 which inactivates GLP-1 - act similarly to GLP-1 but are advantageously available as an oral medication (sitagliptin OD, vildagliptin BD) |
|
|
|
What occurs to the incretin effect in type II diabetes?
|
IT IS DIMINISHED
- Levels of GLP-1 are decreased - Insulinotropic response to Glucose-dependent insulinotropic peptide is diminished but not absent - defective GLP-1 release and diminished response to glucose-dependent insulinotropic peptide may be important factors in glycaemic dysregulation in type 2 diabetes |
|
|
|
Which factors inhibit insulin secretion?
|
>Decreased [glucose]
>Increased [somatostatin] - pancreatic and gastric >Noradrenaline - high [] beta adrenoceptors >Adrenaline - beta adrenergic receptors >Increased [insulin] - paracrine / autocrine feedback |
|
|
|
Which factors stimulate glucagon secretion?
|
>Decreased [glucose]
>Increased [free aa] >Adrenaline >Increased [insulin] |
|
|
|
What causes inhibition of glucagon secretion?
|
>Increased [glucose]
|
|
|
|
What are examples of insulin dysregulation?
|
>Insulinoma - constitutive insulin secretion from a beta cell tumour
>Persistent Hyperinsulinaemia and hypoglycaemia in infancy (PHHI) - mutations in the genes encoding the sulfonylurea receptor and the K+ inward rectifier channel, resulting in abnormal function of ATP sensitive K+ channels, resulting in hyperinsulinaemia - PHHI is the commonest cause of persistent hypoglycaemia in infancy - incidence is 1/50000 births among caucasions, but higher where the parents are consanguinous |
|
|
|
What are two potential issues of insulin administration?
|
1. Insulin induced hypoglycaemia, leading to coma if glucose <2mM
- injection of excess insulin - failure to eat after administration of insulin 2. Antibody response - develops resistance to exogenous insulin |
|
|
|
In what structure is insulin administered?
|
>Hexamer, although active only as a monomer
|
|
|
|
What is the structure of the insulin receptor?
|
>α2β2 heterotetramer
- extracellular α subunits = site of insulin binding - transmembrane β subunits = intrinsic receptor tyrosine kinases (RTKs) - receptor highly expressed in adipose tissue and the liver |
|
|
|
What is the receptor signalling pathway for the insulin receptor?
|
>Activated receptor TyrK phosphorylates Tyr residues on insulin receptor substrate (IRS) 1 and 2
- IRS1 and 2 act as adapter proteins, phosphotyrosine residues interact with src-homology (SH)2 domains in downstream signalling proteins >PI3K inserts GLUT4 >Grb2 |
|
|
|
What decreases plasma [glucose]?
|
>Insulin
- as glucose falls, insulin secretion reduces >Many cells become unable to take up glucose, and switch to alternative fuels for energy e.g. FA >Neurones need constant supply of glucose - glycogen reserves - breakdown stimulated by - absence of insulin and presence of glucagon |
|
|
|
Which is more common, diabetes mellitus or diabetes insipidus?
|
>Diabetes mellitus
|
|
|
|
What is the prevalence of diabetes mellitus?
|
2-5%
|
|
|
|
What is the % of population with prediabetic symptoms in the UK?
|
>>15% of the population
- impaired glucose tolerance |
|
|
|
Which population is most affected by diabetes?
|
>Pima Indians, USA
|
|
|
|
What is the progression of type I diabetes?
|
AGE 5-10 - normal Beta cell mass
- precipitating event - overt immunologic abnormalities - normal insulin release AGE 10-15 - decreasing Beta cell mass - progressive loss of insulin - glucose normal AGE 20-25 - overt diabetes |
|
|
|
What is thought to cause the decrease in beta cell mass in diabetes?
|
>Autoimmune destruction of the beta cells
|
|
|
|
When is diabetes suspected?
|
>May be suspected when random whole blood venous samples show a value in excess of 11.1mM
OR - the fasted 8h sample exceeds 7mM - if 6.1-7mM should do an oral glucose test |
|
|
|
What is given to patients when a diabetes glucose is uncertain?
|
GLUCOSE TOLERANCE TEST IS UNDERTAKEN
>Patient fasts and rests overnight - no smoking allowed >Fasting glucose sample is taken >Glucose solution is given by mouth (75g in 300ml water) >Blood and urine samples are taken after 2 hours |
|
|
|
What is expected from a diabetic patient after a glucose tolerance test?
|
Whole blood:
- Fasting sample: >6.7 - 2h after glucose load: >10 Plasma - Fasting sample: >7.8 - 2h after glucose load: >11.1 |
|
|
|
What is expected from a patient with impaired glucose tolerance (pre diabetic) after a glucose tolerance test?
|
Whole blood:
- Fasting sample: >6.7 - 2h after glucose load: 6.7-10 Plasma - Fasting sample: >7.8 - 2h after glucose load: >7.8-11.1 |
|
|
|
Why is it important that glucose is regulated?
|
>Sustained increases in plasma glucose have serious clinical consequences
>Insulin is required to stimulate post prandial glucose storage - relevant therefore to compare fed and fasted state |
|
|
|
How is insulin synthesised?
|
>Preproinsulin - cleaved to form proinsulin - which is converted by prohormone convertases, activated in the acidified secretory granules, remove the C-peptide (measured in diagnostic tests)
>Insulin is final cleavage product - packaged in golgi into secretory granules which accumulate in the cytoplasm TIMING - 20 minutes to cleave to proinsulin and package in Golgi - 50-140 min to cleave C peptide - >80-180m - insulin forms hexamers with Zn - granule core surrounded by C peptide, explaining the need for zinc |
|
|
|
What does insulin depend upon for secretion?
|
>Ca++ release
|
|
|
|
What configurations does insulin typically form in solution and why?
|
>Tendency to form dimers in solution due to hydrogen bonding between the C termini of B chains
>In the presence of zinc ions, insulin dimers associate into hexamers - monomers and dimers readily diffuse into blood, whereas hexamers diffuse poorly - this phenomenon resulted in the development of recombinant analogs of insulin |
|
|
|
Where is insulin injected?
|
>Into the fat layer just under the skin
- if the needle is injected into the muscle, the insulin absorbed (moved into the bloodstream) too quickly - insulin is absorbed quickest when it is given in the abdomen >Site of injection should be changed regularly - helps prevent changes to skin such as lumps, swollen areas or thickened skin - if skin changes occur in the usual area then the area should be changed |
|
|
|
Which factors impact the different insulin regimens used?
|
NO INSULIN INJECTION REGIMEN SATISFACTORILY MIMICS NORMAL PHYSIOLOGY
>Choice of insulin will depend on many factors including: - age - duration of diabetes - lifestyle (dietary patterns, exercise schedules, school, work commitments) - targets of metabolic control and particularly, individual patient / family preferences) >At least two injections of insulin per day are advisable in most children - occasionally, particularly during the partial remission phase in younger children, one injection per day maintains satisfactory glycaemic control >Most regimens include a proportion of soluble short acting or rapid acting insulin analogues, but some young children or those in the partial remission phase maintain satisfactory metabolic control on intermediate or long acting insulins alone |
|
|
|
Name three typical regimens of insulin.
|
1. Two injections daily
- of a mixture of short and intermediate acting insulins (before breakfast and the main evening meal 2. Three injections daily - using a mixture of short and intermediate acting insulins before breakfast, short acting insulin alone before an afternoon snack or main evening meal, intermediate acting insulin before bed or variations of this 3. Basal bolus regimen - of short acting insulin 20-30 min before main meals (e.g. breakfast, lunch and the main evening meal), intermediate or long acting insulin at bedtime 4. Basal bolus regimen of rapid acting insulin analog immediately before main meals (e.g. breakfast, lunch and main evening meal) intermediate or long acting insulins at bedtime) |
|
|
|
What are the characteristics of short acting insulin?
|
>Short-acting (soluble, regular) insulin is used as an essential component of most daily replacement regimens either
- in combination with intermediate acting insulin in a twice daily regimen - as pre-meal bolus injections in basal-bolus regimens (20-30 before meals) >Soluble is the only insulin suitable for IV therapy >Soluble insulin is used in the following crisis situations - diabetic ketoacidosis - control of diabetes during surgical procedures - hyperglycaemic episodes at home (e.g. during intercurrent illness) |
|
|
|
What are the characteristics of rapidly acting insulin analogs?
|
>Several novel insulin analogues are being developed
- two rapid acting monomeric types are currently available for children - they have rapid onset and shorter duration of action than soluble insulin >Rapid acting analogues can be given immediately before meals because there is evidence that the rapid action not only reduces postprandial hyperglycaemia but that postprandial and nocturnal hypoglycaemia may also be reduced - in selected children they offer the useful option of being given after food to toddlers who are reluctant to eat - may also be used during sick days with hyperglycaemia and potential ketosis - most often used as prandial or snack boluses in combination with longer acting insulins given twice or more times daily |
|
|
|
What are the characteristics of intermediate acting insulin analogs?
|
>The action profiles of these insulins make them suitable for twice daily regimens and for pre-bed dosage in basal-bolus regimens
>Two principal preparations are used: - isophane NPH insulins - crystalline zinc acetate insulin (insulin zinc suspensions) or lente insulins >Isophane insulins are extensively used in children mainly because of their suitability for mixing with soluble or rapid acting insulins in the same syringe, vial or cartridge without interaction - when soluble insulin is mixed with lente preparations, it reacts with excess zinc, blunting its short acting properties |
|
|
|
What are the characteristics of long acting insulin?
|
>Ultralente and ultratard insulins were designed to have a duration of action of more than 24h to meet basal insulin requirements and therefore could be used in basal bolus injection regimens
- their action profile in children appears to be extremely variable and they may have to be injected twice daily to meet basal insulin requirements |
|
|
|
What are pre-mixed insulin preparations?
|
>Fixed ratio mixtures of soluble and isophane
- popular in prepubertal children on twice regimens - reduce errors in drawing up insulin - remove flexibility offered by separate adjustment of the two types >No evidence that pre mixed insulins are less effective - some evidence of poorer metabolic control when used in adolescents >Pre mixed insulins are most commonly used in pen injector devices - useful when compliance or adherence to the regimen is a problem |
|
|
|
What are some key facts about obesity?
|
>Most common nutrition-related disorder in the Western world
- around half of UK adults are overweight or obese >Becoming an increasing problem in developing nations as well - highest rates occurring in the poor and undereducated >Significantly reduces life expectancy - associated with an increased risk for several conditions, such as Type II diabetes, coronary heart disease and cancer >Adipose tissue was seen as a way of storing large amounts of Triacylglycerol - an energy store - now recognised as an important endocrine tissue - secretory profile disturbed in obesity |
|
|
|
What are increased risks in the obese?
|
BMI > 30Kg/m^2 associated with
- type II diabetes - hyperinsulinaemia - glucose intolerance - hypertension and stroke - CHD - some cancers >Often suffer from metabolic syndrome - increasing risk for CAD, stroke and type II diabetes - risk factors include central obesity, insulin resistance, high BP, dyslipidaemia, hyperglycaemia, mild chronic inflammation |
|
|
|
What are the genetic factors involved in obesity?
|
>Heritability of obesity is extremely high
- genetic variation can also affect numours obesity related phenotypes 1. appetite regulation 2. fuel metabolism 3. body weight distribution 4. risks associated with obesity >Our current environment (high calorie food and sedentary lifestyles almost guarantee that any propensity towards obesity will become manifest |
|
|
|
What are examples of genetic mouse models of obesity?
|
1. LETHAL YELLOW MUTANT MOUSE (AY)
- expresses agouti protein, antagonising hypothalamic neurones - lethal to homozygotes - heterozygotes have yellow coat, mature onset obesity, type II diabetes, hyperleptinaemia, tumour susceptibility 2. OBESE MOUSE (ob/ob) - does not express the product of the ob gene - leptin - mutant mice gain weight rapidly, to become 3x the size of control mice - uncontrollable food intake, obesity, type II diabetes, insulin resistance and hyperinsulinaemia 3. DIABETIC OBESE MOUSE (db/db) - does not express the db gene - leptin receptor long form - mutant mice are larger / obese relative to heterozygous littermates by one month, with increased fat deposition and hyperglycaemia by 8 weeks 4. FAT MOUSE (fat/fat) - obesity develops relatively slowly, increased fat deposition, hyperproinsulinaemia but not hyperglycaemia, not prone to diabetes |
|
|
|
In what ways can obesity be diet induced in animal models?
|
>High fat diet
- 58% of kcals from fat, 25.6% carb, 16.4% protein - mice fed on this diet exhibit increased weight gain, modest hyperglycaemia, insulin resistance etc, used as a model of impaired glucose tolerance and early type II diabetes - when fed to different strains of mice, some strains are more resistant to the diet induced obesity than others, or to particular risk factors >Cafeteria diet - standard chow and assortment of human snack items - encourages hedonic feeding - CAF fed consumed 30% more calories and gained the most weight (c.f. HFD) - worse hyperglycaemia, highest FFAs, higher circulating inflitrating MCV, dramatically altered pancreatic islets - better model of metabolic syndrome |
|
|
|
What is the traditional view of adipose tissue?
|
ADIPOSE TISSUE SEEN AS A WAY OF STORING ENERGY FOR LATER USE
FED STATE: - insulin stimulates the uptake of glucose, which is used in the synthesis of TAG - TAG stored in large droplet for later use DURING FASTING / EXERCISE: - low insulin / adrenaline stimulates lipolysis of the stored TAG - the released NEFA enter the plasma as fuel for other tissues |
|
|
|
How does insulin affect adipocytes?
|
>PI3 kinase pathway mediates many of the more immediate responses to insulin, such as
- GLUT4 translocation - glycogenesis - inhibition of lipolysis - shows less activity in insulin resistance >MAPK pathway mediates the proliferative mitogenic effects of insulin such as cell proliferation - unaffected by insulin resistance - in hyperinsulinaemia, this pathway may have atherogenic and cancerogenic effects, which could be further exacerbated by insulin administration |
|
|
|
What the function of brown adipose tissue?
|
>Produces heat via non shivering thermogenesis
>Upon stimulation by Nad B3 adrenoceptors, lipolysis and FAO are activated - proton gradient generated by the electron transport chain is then wasted via UCP1 >Brown differs from white adipose tissue in several respects - the TAG droplets are multilocular - mitochondria are larger and higher in density - highly innervated by SNS and capillary network is denser BAT thought to be significant only in infants, has now been confirmed in adults, particularly after cold acclimation |
|
|
|
What is the adipose tissue development pathway?
|
1. Stem cell
- pluripotent 2. Pre adipocyte - appears similar to SC but committed to adipocyte, produces leptin but no other adipokines 3. Mature adipocyte - expresses enzymes for lipid transport synthesis etc, insulin receptors and senstivity - makes adipokines such as leptin, chemerin, adiponectin |
|
|
|
What is PPARy and why is it important for adipose tissue development?
|
>Transcription factor - necessary for adipogenesis
- increase associated with smaller adipocytes, less ectopic deposition in muscle and liver and improved insulin sensitivity - also needed for maintenance of cell differentiation >Thiazolidinediones are synthetic ligands to PPARy and have been shown to improve insulin sensitivity, lower plasma glucose, and alter the secretory profile of adipose tissue away from the proinflammatory direction |
|
|
|
What are lipodystrophies?
|
>Clinical disorders often involve lipoatrophy, or selective loss of adipose tissue from particular anatomical regions
- may be genetic or impaired >Patients often have aspects of metabolic syndrome e.g. insulin resistance, dyslipidaemia, hypertension >Some of the symptoms seen in lipodystrophies can be alleviated by the administration of adipocytokines, such as leptin or adiponectin - lipodystrophies highlight the benefits of adipose tissue |
|
|
|
What might occur in a PPARy mutation?
|
1. LIPODYSTROPHIES
2. OBESITY >PPARy2 stimulates the differentiation of pre-adipocytes to adipocytes - overactive mutants may result in greater differntiation of adipocyte - affected had substantially greater obesity than the rest of the test subjects, BMI of up to 47.3, but lower fasting insulin levels |
|
|
|
What is the adipose tissue development pathway?
|
>Portal theory
- anatomical location of visceral adipose tissue is such that its products drain directly into the portal vein, to the liver - VAT is hyper lipolytic, resistant to insulin signalling, sensitive to adrenaline, therefore releases more fatty acids - this affects liver metabolism by interfering with insulin signalling (possibly via PKC), liver insulin resistance - insulin resistance in the liver causes it to increase production of VLDL and glucose - excess visceral fat might also be a market that the subcut fat to act as an energy sink has been excluded |
|
|
|
Why is adipocyte size important?
|
>Can increase diameter x20, hypertrophy associated with
- proinflammatory adipokines - desensitizing to insulin - more cell death, recruiting more MCV |
|
|
|
What are adipokines?
|
>Adipokines are released from a huge number of adipokines (>50) most of which are proinflammatory
- now believed that many of the conditions associated with obesity are mediated by the changes in adipokine secretion that occur as obesity progresses - adipokines may act at the autocrine and paracrine levels with both central and peripheral effects - adipokine levels have now been shown to be linked to a wide range of conditions, such as hypertension, cardiovascular disease, insulin resistance etc |
|
|
|
From where is leptin secreted? Where does it act?
|
>Leptin is a protein secreted primarily by mature adipocytes
- receptors are concentrated in the feeding centres of the hypothalamus, but also in peripheral tissues |
|
|
|
Which factors affect leptin secretion?
|
>Increased body fat, but also fluctuates in response to feeding (in response to insulin), increases with overfeeding and decreases with fasting
|
|
|
|
What are the functions of leptin?
|
>Decrease food intake and increase energy expenditure
|
|
|
|
Obese individuals have high levels of leptin, why does leptin not regulate their body fat?
|
>Leptin crosses the blood brain barrier via a saturable transport mechanism that probably involves a short soluble variant of the leptin receptor
>The levels of leptin found in cerebrospinal fluid of obese individuals is relatively low compared to the high plasma levels, central administration does have some effect on weight loss - a fault in downstream signalling could also be involved - dietary fat and fructose (which do not stimulate insulin production) do not increase leptin secretion, this may be one reason why such diets are associated with obesity |
|
|
|
What are the immune effects of leptin?
|
>Normal immune function is suppressed during nutritional deprivation, but this can be reversed by the administration of leptin
>Also a chemoattractant - they high levels secreted by adipocytes in obesity may be responsible for the infiltration of MCV into adipose tissue >Leptin also has an effect on the reproductive system |
|
|
|
What is adiponectin?
|
>Normally found at very high levels in the plasma
- only secreted by mature adipoytes - plasma levels are lower in obesity >Expression depends on PPARy, but is inhibited by TNF-alpha, also lower in large adipocytes - low levels associated with hyperinsulinaemia, insulin resistance and future risk of type II diabetes >Several gene polymorphisms have been associated with increased risk of metabolic syndrome or type II diabetes >Levels are increased by weight loss, exercise and synthetic ligands to PPARy, such as thiazolidinediones |
|
|
|
What is the effect of increasing adiponectin levels?
|
>Increasing adiponectin levels increases insulin sensitivity
>Muscle has receptor AdipoR1, adiponectin causes an increase in glucose uptake and increases beta oxidation, which is most likely the cause of the decrease in muscle lipid stores - increasing insulin sensitivity >Liver (AdipoR2) also has improved insulin senstivity in response to adiponectin, decreasing liver gluconeogenesis and plasma glucose >Adiponectin is also antiinflammatory, inhibiting TNF-alpha secretion by monocytes and MCV >Higher adiponectin levels are also associated with a decreased risk for cardiovascular problems |
|
|
|
How is obesity associated with inflammation? Via which markers?
|
>Associated with mild but chronic inflammation
- may be due to hypoxia in expanding adipose tissue, or high levels of FFA stimulating toll like receptors >In obesity, many markers of inflammation are at higher levels in the plasma, including C reactive protein, IL-6, serum amyloid A (SAA) etc. >Adipose tissue produces - IL-6, 8, Ibeta, TNF alpha, chemokines - such proteins stimulate immune responses, recruiting immune cells and stimulate maturation of immune cells - these proteins inhibit preadipocyte maturation - weight loss leads to decreased levels of these proteins |
|
|
|
What antiinflammatory proteins are produced less as obesity increases?
|
>IL-10 and adiponectin
- inhibits MCV activation and stimulates them to produce IL-10 |
|
|
|
Which proinflammatory proteins are produced in response to obesity?
|
>Leptin, IL-6, angiotensin
- all increased in plasma in obese >IL-6 predicts onset of T2DM - causes liver to make inflamm proteins e.g. CRP |
|
|
|
What is meant by MCV infiltration in obesity?
|
>MCV infiltration of adipose tissue is increased in obesity
- MCVs are typically clustered around individual adipocytes - forming crown like structures (CLS) >CLS always found around dead adipocytes, that appear to have undergone necrosis rather than apoptosis - adipocyte death was increased with the size of the adipocytes, even in lean individuals, or mice models of adipocyte hypertrophy without obesity >Infiltrating MCV may then secrete large amounts of proinflammatory cytokines - infiltration has been shown to occur before hyperinsulinaemia |
|
|
|
What are the proposed causes of insulin resistance?
|
>May be due to inhibition of the signalling pathway, particularly caused by inflammatory cytokines, can lead to Type II diabetes mellitus
- Adiponectin improves insulin sensitivity, as does leptin, but in obesity, adiponectin levels are lower, and there is leptin resistance - Adiponectin is anti-inflammatory and has effects on muscle and liver metabolism |
|
|
|
Why is there an increased risk of hypertension in obesity?
|
>A risk factor for cardiovascular disease
>Most adipokines (including inflammatory cytokines and leptin) act by altering the behaviour of endothelial cells (decreasing NO production, increasing production of vasoconstrictors) and smooth muscle cells (increasing proliferation) - Adipose tissue also produces components of the renin- angiotensin system, which act to increase blood pressure >Adiponectin increases NO production, and decreases SMC proliferation, as well as reducing oxidative stress - Also inhibits atherogenesis, partly due to opposing the actions of inflammatory cytokines |
|
|
|
Why is there an increased risk of certain cancers in obesity?
|
>Obesity is associated with, for some cancers, increased incidence, increased aggression of tumours, increased recurrence and increased mortality
>Adipokines may be involved acting via endocrine or paracrine mechanisms - Some adipokines act as growth factors for cancer cells and they may promote angiogenesis (e.g. adipose tissue may produce Vascular Endothelial Growth Factor) normally needed for growth of adipose tissue >Adipose tissue appears to produce substances that also favour metastasis, such as Hepatocyte Growth Factor >Adiponectin levels correlate inversely with cancer risk and progression, suppresses proliferation and angiogenesis, and favours apoptosis |
|
|
|
What are the characteristics of type I diabetes?
|
>Juvenile onset diabetes, IDDM
>Caused by autoimmune destruction of β cells >Age of onset 1-25 years >Lean body physique >0.5% prevalent >~50% probability of inheritance >Treated with insulin injections |
|
|
|
What are the characteristics of type II diabetes?
|
>Maturity onset diabetes, type II diabetes
>Defective insulin secretion and insulin resistance >Age of onset >40 years >Obese physique >2% prevalent >~70-80% risk of inheritance >Treated with diet, drugs |
|
|
|
What is the initial pathophysiology of type I diabetes?
|
COMBINATION OF FACTORS
1. Genetic predisposition - HLA linked genes and other genetic loci 2. Environmental insult - viral infection: molecular mimicry and or damage to beta cells TRIGGERS >immune response against normal beta cells, AND/OR >immune response against altered beta cells LEADING TO >Autoimmune attack - beta cell destruction = type I diabetes |
|
|
|
How do histological changes in type I diabetes differ from those in type II?
|
1. Type I
- autoimmune reaction against beta cells leads to infiltration of the pancreatic islet by T cells - eventually destroys the gland 2. Type II - pancreatic islet is not usually affected - however in some patient with longstanding diabetes, replacement of some of the islet by amyloid can be seen - diabetic amyloid is composed of amylin fibres derived from beta cells |
|
|
|
What are the short term effects of lack of insulin in type I diabetes?
|
1. Glucose transport and utilisation reduced
- hyperglycaemia 2. Plasma glucose increases more because gluconeogenesis and glycogenolysis is not suppressed by insulin 3. Breakdown of fat (lipolysis) not suppressed by insulin 4. Protein breakdown increased - more free amino acids, stimulating glucagon further |
|
|
|
What are the consequences of the metabolic disturbances in type I diabetes?
|
INSULIN DEFICIENCY leads to
1. decreased tissue glucose utlisation - spillover into blood 2. increased protein catabolism and increased free amino acids 3. adipose tissue breakdown (FFAs) INCREASED GLUCAGON SECRETION become excessive (unregulated by insulin) leading to - gluconeogenesis - ketogenesis these effects lead to POLYPHAGIA (lipolysis) KETOACIDOSIS (causing diabetic COMA, ketonuria, and polyuria/polydipsia) HYPERGLYCAEMIA (glycosuria - polyuria and polydipsia) |
|
|
|
What are the effects of insulin and glucagon on glucose uptake in muscle?
|
INSULIN: increases
GLUCAGON: No effect |
|
|
|
What are the effects of insulin and glucagon on glucose uptake into the liver?
|
INSULIN: No effect
GLUCAGON: No effect |
|
|
|
What are the effects of insulin and glucagon on glycogen synthesis?
|
INSULIN: Increases
GLUCAGON: Decreases |
|
|
|
What are the effects of insulin and glucagon on glycogenolysis?
|
INSULIN: Decreases
GLUCAGON: Increases |
|
|
|
What are the effects of insulin and glucagon on glycolysis?
|
INSULIN: Increases
GLUCAGON: Decreases |
|
|
|
What are the effects of insulin and glucagon on gluconeogenesis?
|
INSULIN: Decreases
GLUCAGON: Increases |
|
|
|
What are the effects of insulin and glucagon on lipolysis?
|
INSULIN: Decreases
GLUCAGON: Increases |
|
|
|
What are the effects of insulin and glucagon on ketosis?
|
INSULIN: Decreases
GLUCAGON: Increases |
|
|
|
What are the effects of insulin and glucagon on lipogenesis?
|
INSULIN: Increases
GLUCAGON: Decreases |
|
|
|
What are the effects of insulin and glucagon on uptake of uptake of amino acids into muscle / protein synthesis?
|
INSULIN: Increases
GLUCAGON: Decreases |
|
|
|
What is the mechanism of insulin resistance?
|
CAUSED BY MULTIPLE RISK FACTORS:
1. unbalanced diet, overeating - causing fat accumulation in internal organs (exacerbated by shortage of exercise also leading to 2. low blood flow in skeletal muscle, dysfunction of the liver (exacerbated by hyperlipidaemia) 3. aging also a risk factor 4. decreased insulin secretion - leading to elevated blood sugar levels also 5. Inflammation of periodontal tissues caused by oral bacterial infection |
|
|
|
What is the pathogenesis of type II diabetes?
|
OFTEN REVEALED BY LONG TERM COMPLICATIONS
1. Multiple genetic defects - primary beta cell defect, deranged insulin secretion - peripheral tissue insulin resistance 2. Environment - obesity - peripheral tissue insulin resistance - inadequate glucose utilisation LEADING TO - hyperglycaemia, which exhausts β cells |
|
|
|
What are the complications of diabetes?
|
>Increased incidence of cardiovascular disease (M.I)
>More peripheral vascular disease >Renal disease (microangiopathy) >Retinal disease (microangiopathy) >Lens opacity (cataracts - via crystallin glycation) >Neuropathy (impaired nerve conductance e.g. impotence) >Skin infections (gangrene, thrush and other yeasts) >Osteoarthritis |
|
|
|
How does glucose affect haemoglobin? Why is this clinically useful?
|
>Excess glucose causes glycation of proteins
- therefore glycation of an easily accessible protein such as haemoglobln gives a good indication of the success of control of glucose levels with insulin or drug treatment - HbA1c may be measured |
|
|
|
What is Amadori rearrangement?
|
It is a critical stage in the glycation of proteins in hyperglycaemia
- irreversible conversion of a Schiff base to a ketoamine |
|
|
|
What occurs to the basement membrane in diabetics? How does this effect the kidney?
|
>Thickens, and increased permeability to blood proteins
- results in nephropathy >Macroscopically this results in a granular surface - extensive sclerosis of cortical glomeruli - cut surface shows destruction of the renal papillae and scarring consistent with previous attacks of pyelonephritis >Microscopic appearance of diabetic nephrophathysecondary to microvascular injury - AGE lead to biochemical abnormalities of the basement membrane and the mesangium and accumulation of mesangial matrix - this could diffusely involve all of the glomerular basal membrane or could be focal/nodular as seen here (Kimmelstiel-Wilson nodules) - accordingly the changes are described as diffuse or nodular glomerulosclerosis |
|
|
|
Via what mechanism does glycation cause an increase in free radicals?
|
>Glycation of SOD causes it to lose activity
- converts O2.- and H+ to H2O2 and H2O |
|
|
|
Why do infections commonly occur in diabetics?
|
>High levels of glucose make a rich medium for organisms infecting the skin
>Especially when small blood vessels are damaged and when you cannot feel the discomfort because of neuropathy - in some cases neuropathy can cause intense pain |
|
|
|
What causes sorbitol and fructose to accumulate in cells? What is the consequence of this?
|
>High glucose levels
>Causing osmotic effects which may damage cells such as lens cells and nerve cells |
|
|
|
What causes retinopathy in diabetes?
|
>Glycation of basement membrane protein of capillaries - increased permeability - bleeding
|
|
|
|
What causes lens opacity in diabetes?
|
>Glycation of crystallins, the clear proteins of the lens
>Accumulation of polyols |
|
|
|
What causes neruopathy in diabetes?
|
>Changes in small blood vessels due to glycation can contribute to neuropathy
>Possible accumulation of polyols >Disturbance of inositol metabolism, signal transduction issues |
|
|
|
What causes glomerular abnormality in diabetes?
|
Mainly due to changes in basement membrane
|
|
|
|
What causes osteoarthritis in diabetics?
|
>Poor glucose control can lead to modification of collagen and other structural proteins so that they cannot be readily degraded, enhanced by radical induced damage
|
|
|
|
What causes cardiovascular disease in diabetics?
|
1. Activation of factor VII (due to high VLDL)
2. Impaired endothelial function - i.e. reduced release of nitric oxide, therefore greater arterial contraction and reduced blood flow 3. Modification of lipoproteins - macrophage cells have receptors for AGE proteins (RAGE), which lead to the uptake and accumulation of glycated LDL, and therefore cholesterol in the atherosclerotic plaque 4. Proliferation of smooth muscle cells, part of development of atherosclerosis due to activation of MCV and release of chemokines and growth factors from macrophages 5. Hyperinsulinaemia seen in some TIIDs may also have independent effects which enhance atherosclerosis |
|
|
|
What are possible future therapies for diabetes?
|
>Existing and emerging therapies aimed at regulating the autoimmune response largely involve broad based immunoregulatory strategies
- inhibition or deletion of lymphocyte subsets - reestablishment of immune tolerance, via activation of Tregs e.g. nonmitogenic antiCD3 or antithymocyte globulin >Some have shown efficacy although there are risks with broad immunologic approaches - research hampered by lack of biomarkers |
|
|
|
What are the characteristic features of diabetes mellitus?
|
>Lack of insulin action means glucose is not taken up and stored in muscle, liver and fat which causes increased circulating blood glucose levels
>Muscle, liver and fat turn to other energy sources such as fatty acid - increases production of acetyl CoA - condense into ketone bodies which acidify blood (ketoacidosis) >High circulating levels of glucose eventually overcome the ability of the kidney's glucose reabsorption system so that glucose spills into urine >High glucose levels in the urine combined with a high excretion of H+ as a result of the acidity water flows into the bladder >When present at high levels in the circulation the glucose can covalently link to proteins in the blood or on the walls of blood vessels, a process known as glycation |
|
|
|
What are the two major antibodies found in the circulation of type I diabetics?
|
>To insulin and to glutamic acid decarboxylase
|
|
|
|
What are MHC II alleles?
|
>MHC II is a cell surface receptor consisting of 4 subunits
- found on the surface of certain antigen presenting cells where its role is to present antigens to helper T cells - diversity arises as we have three alleles for this gene (DP, DQ and DR), and at each there are several copies of the genes coding for the 4 subunits - these are highly polymorphic i.e. even at a given allele, there is a great deal of sequence variation in the general population >Therefore there are a huge number of possible combinations - each wil have a specific peptide binding preference and thus will identify different antigens - estimated that between 20 and 50% of all type I diabetes might be variants of the MHC II locus |
|
|
|
Which MHC II alleles are associated with a higher risk of type I diabetes?
|
>DQA*301 or DRB1*401
>33% of diabetics have copies of both alleles (3/4 genotype) - very rare in non diabetics >57% of non diabetics lack one or another allele >MHC II protein make individuals susceptible to autoimmune destruction of pancreas - mechanism poorly understood |
|
|
|
What other genes have been associated with type I diabetes?
|
>Polymorphisms in the non coding regions of the insulin gene
>IGF-2 >FGF (fibroblast growth factor) |
|
|
|
What are possible non insulinergic treatments for type I diabetes?
|
>Islet transplantation - some success, but limited by availability of the islets
- artificial islets are some way off >Immunosuppressive therapy of those at risk (in trial) >Prophylactic injections of insulin in those at risk of diabetes seems to delay onset of diabetes >Innoculation against factors that cause autoimmunity |
|
|
|
What is the molecular basis of insulin resistance?
|
1. Arises initially in muscle and fat, then at later stages in the liver
2. Insulin resistance observed in patients without frank diabetes, but who have the metabolic syndrome - minority of these patients progress to full diabetes 3. Lower pancreatic output of insulin with insulin resistance and inability of insulin to suppress gluconeogenesis in the liver - may be needed to precipitate TIID |
|
|
|
Which factors inducing the resistance of insulin have been investigated? Which other diseases can these arise in?
|
Include:
- elevated fatty acids - inflammatory mediators - high levels of hormones that antagonise insulin (glucagon, adrenaline, glucocorticoids) - high levels of insulin >May arise in obesity, metabolic syndrome and a number of endocrine disorders e.g. Cushing's syndrome |
|
|
|
How does insulin resistance affect glucose uptake / release in muscle, adipose and the liver?
|
FAT AND MUSCLE
- reduced uptake of glucose via GLUT4 transporters LIVER - gluconeogenesis unsuppressed therefore increased hepatic release of glucose occurs - insulin doesn't normally influence glucose uptake in the liver but normally antagonises the effects of glucagon in the normal liver and suppresses gluconeogenesis and stimulates storage of glucose as glycogen |
|
|
|
What is the effect of insulin resistance on lipid metabolism?
|
>In adipose tissue, insulin normally suppresses lipolysis and promotes storage of fat
IN RESISTANCE - there is increased release of fatty acids into the circulation and then reconversion of these acids by the liver into TAG which are recirculated as VLDL in high concentrations (dyslipidaemia) - high levels of insulin have other consequences - e.g. stimulation of some cells of the sympathetic nervous system leading to hypertension - in some cases it may increase androgen production by ovaries which contributes to the hormonal disturbances seen in polycystic ovary syndrome - including masculinisation |
|
|
|
When might a TIID require insulin?
|
>In TIID at first insulin concentrations might seem normal or high
- in many, but not all cases insulin release decreases with time - at this stage patients begin to require insulin therapy |
|
|
|
What are IRS?
|
Insulin response substrates are important ligands in the insulin response of human cells:
- 4 types, but key two are IRS1 and 2 - IRS1 is the main type that leads to the metabolic effects of insulin - IGF-1 secreted from the liver in response to stimulation from growth hormone, has similar growth stimulating properties to insulin, works through IRS 2, IGF-1 has its own receptor but this like insulin also activates IRS-1 and 2 - Activation of IRS-1 and 2 is linked to the growth function and is active in the beta cells of the pancreas and other cell types Insulin has other effects related to growth and development |
|
|
|
What are suggested to be the genetics of type II diabetes?
|
>Genetic studies indicate several genes, i.e. a polygenic disease
- no clear pattern of inheritance >No direct evidence that glucose transporters are involved >Indications that some individuals with insulin resistance have one of several mutations in IRS1 genes and IRS2 genes - does not explain all of TIID >Diverse genetic defects associated with the complex signalling mechanisms surrounding insulin >Some identified because they are found in young patients and patients with NIDDM characteristics - Different from middle aged patients with mild genetic phenotypes does not precipitate diabetes without another event or late appearance of a clinical condition |
|
|
|
What is MODY?
|
MATURITY ONSET DIABETES OF YOUNG
>Around 2% of diabetes - 0.1% of population >Similarities to TIID - no islet cell antibodies - beta cell destruction >Differences from TIID - patient's age - autosomal dominant pattern of inheritance - no association with obesity >Genes responsible have been identified in some pedigrees |
|
|
|
Describe two different types of MODY.
|
MODY:
1. Mutations in Hepatic Nuclear Factor 4 alpha - cause 1% of MODY - insulin secretion normal at birth, decreases dramatically with age 2. Mutations in glucokinase, affects glucose sensing in pancreas - 15% of MODY - Insulin secretion impaired at birth, but remains relatively stable 3. Mutations in Hepatic Nuclear Factor 1 alpha - Causes 70% of MODY - insulin secretion normal at birth, decreases dramatically with age 4. PDX-1 transcription factor in beta cells involved in regulating insulin gene expression - estimated to cause 1% of MODY 5. HNF 1 beta transcription factor - estimated to cause 1% of MODY X. Estimated to cause 11% of MODY |
|
|
|
What is mitochondrial diabetes?
|
>Occurs in families with an heritable form of diabetes
>Diabetes exclusively inherited from mother - level of penetrance is variable >Caused by mutations in mitochondria (hence maternal transmission) >Accounts for 1-2% of all diabetes >Often misdiagnosed as either type 1 or type 2 diabetes >When mutant mitochondria predominate, oxidative capacity is reduced - in beta cells this results in a reduced capacity to secrete insulin in response to glucose as insulin secretion is linked to glycolysis >Symptoms e.g. muscle weakness, deafness, neurological problems and lactic acidosis |
|
|
|
Which other (rare) conditions have similar symptoms to TIID?
|
>Antibodies to the insulin receptor or insulin
>Mutations in insulin receptor (these can cause leprechaunism) >Glucagonomas |
|
|
|
What other endocrine related effects may stimulate diabetes or related symptoms?
|
1. Hyperthyroidism
- increases insulin resistance and diabetes in hyperthyroid patients - unclear why this is the case 2. Acromegaly and growth hormone - 60% acromegalics insulin resistant and 20% diabetic - GH a counterregulatory hormone - administration of GH in normal subjects increases insulin resistance - GH deficient children are hypoglycaemic 3. Glucocorticoids - excess glucocorticoids - endogenous tumour or exogenous - increases insulin resistance and frank type II diabetes - beta cell function normal - diabetes is usually reversible if steroid levels are reduced - mechanism: IRS1 decreases tyrosine phosphorylation 4. Adrenaline - Increases insulin resistance and blocks insulin secretion - phaeochromocytoma or exogenous administration - increases glycogenolysis and glucose clearance - decreases tyrosine phosphorylation of IRS 1 |
|
|
|
What are the available treatments for TIID?
|
>Diet and exercise
- difficult to enforce and only a temporary solution >Insulin - recombinantly produced insulin plus variants of insulin designed for fast release >Sulfonylureas increaes insulin secretion >Biguanides e.g. metformin - act on liver mainly and increase response to insulin >Thiazolidinediones - a new class of drug which binds to transcription factor PPARy - mainly thought to act in fat cells, somehow reduces insulin resistance |
|
|
|
What are the key and related structures of the pituitary gland?
|
>Pituitary gland protected at the base of the brain in the sphenoid bone connected by the pituitary stalk
KEY STRUCTURES include: - third ventricle - hypothalamus - pituitary stalk - portal vessels - anterior and posterior lobes |
|
|
|
What are the functional connections between the pituitary and the hypothalamus?
|
>Hypophyseal portal circulation
>Supraoptic hypothalamic tract NB. connected physically to the hypothalamus by the pituitary stalk |
|
|
|
What are the alternative names for the anterior and posterior pituitary gland?
|
>Anterior = Adenohypohysis
>Posterior = Neurohypophysis |
|
|
|
What controls the posterior lobe of the hypothalamus?
|
NEUROHYPOPHYSIS controlled by magnocellular neurones
- cell bodies in paraventricular nuclei (PVN) and supraoptic nucleus (SON) of hypothalamus - axons project into the posterior pituitary lobe along the supraoptic hypothalamic tract FUNCTION - hormones secreted from posterior pituitary gland (oxytocin and vasopressin / ADH) synthesised in PVN and SON then transported along axons of magnocellular neurones to be secreted from posterior pituitary into inferior hypophyseal circulation |
|
|
|
What controls secretions of the anterior lobe?
|
>Secretions of anterior lobe controlled by neuronal factors secreted from the hypothalamus into the hypophyseal circulation
- hypothalamic releasing hormones - hypothalamic inhibitory factors |
|
|
|
Where are the neuronal factors controlling the anterior lobe secretions synthesised?
|
>In parvicellular neurones
- arise in the hypothalamic nuclei in lateral wall of third ventricle and terminate in external layer of median eminence - secreted into hypophophyseal portal veins and delivered to anterior pituitary lobe |
|
|
|
What are the five cell types and 6 hormones of the anterior pituitary?
|
1. Somatotrophs
- GH 2. Mammotrophs - prolactin 3. Gonadotrophs - LH and FSH 4. Corticotrophs - ACTH 5. Thyrotrophs - TSH |
|
|
|
What is the endocrine action of TSH?
|
>Stimulates synthesis and secretion of thyroid hormones
|
|
|
|
What is the endocrine action of Gonadotrophs?
|
>LH and FSH
- stimulate steroid biosynthesis and germ cell maturation in the gonads |
|
|
|
What is the endocrine action of corticotrophs?
|
>Stimulates steroid biosynthesis in the adrenal cortex
|
|
|
|
What is the endocrine action of somatotrophs?
|
>Stimulates growth via IGF-1
|
|
|
|
What is the endocrine action of lactotrophs?
|
>Stimulates lactation
|
|
|
|
What are the structures of TSH, LH and FSH?
|
>Each heterodimeric glycoproteins
- 1 alpha and one beta subunit - alpha subunits are identical, beta subunits differ and confer specificity |
|
|
|
What is the structure of ACTH?
|
>39 amino acid peptide produced by post translational processing of POMC
- other POMC products include MSH and beta endorphin |
|
|
|
What is the structure of prolactin and growth hormone?
|
>approx 190 aa peptides with internal disulphide bonds at equivalent positions
- work through structurally homologous receptors and are capable of stimulating each other's receptors to a limited degree |
|
|
|
How is the anterior pituitary function controlled?
|
>Synthesis and secretion of adenohypophyseal hormones stimulated by hypothalamic releasing hormones
- relatively small simple peptides synthesised in cell bodies of hypothalamic parvicellular neurones 1. Thyrotrophin Releasing Hormone - 3 aas - target thyrotrophs, also stimulate prolactin release from lactotrophs 2. GnRH - 10 aas - stimulate LH and FSH 3. CRH - 41 aas - Corticotrophs - ACTH 4. GHRH - 44 aas - stimulate somatotrophs - GH |
|
|
|
Through which second messengers do releasing hormones work?
|
>GnRH and TRH work via PLA/IP3?Ca++
>CRH and GHRH act via cAMP |
|
|
|
Which two hormones control the synthesis and secretion of growth hormone?
|
Hypothalamic release factor GHRH and hypothalamic inhibitory factor somatostatin (14aas, arising in hypothalamus)
|
|
|
|
What may inhibit GHRH and GnRH from the hypothalamus?
|
>Higher brain centres and stress
|
|
|
|
What is the releasing factor for prolactin?
|
>None yet identified
- appears to be under dominant negative control :. decreased inhibition is stimulus to prolactin synthesis and secretion - major inhibitory factor for prolactin appears to be dopamine (secreted from hypothalamic neurones arising in the arcuate nucleus) |
|
|
|
How is stimulus to the anterior pituitary's function largely controlled?
|
SHORT LOOP NEGATIVE FEEDBACK
>from the anterior pituitary itself >E.g. - Gonadotrophins decrease frequency and amplitude of GnRH pulses from median eminence - GH suppresses subsequent synthesis and secretion of GHRH NB. hormonal signals arising outside hypothalamo-pituitary complex also control synthesis and secretion of peptide and glycoprotein hormones from anterior pituitary |
|
|
|
What is the HPT?
|
>Hypothalamo-Pituitary-Thyroid axis
- TRH stimulates TSH which stimulates T4 and T3 >The hormones released as a consequence of TRH stimulus all inhibit previous stimulating hormones including TRH - negative feedback |
|
|
|
What is the structure of the HPG axis?
|
>Hypothalamus - Pituitary - Gonads
- GnRH, LH and FSH, Progesterone, Testosterone, Oestradiol |
|
|
|
What is the structure of the HPA axis?
|
>Hypothalamus - Pituitary - Adrenal Glands
- CRH, ACTH, Cortisol |
|
|
|
What is an example of hypothalamo-adenohypophyseal override?
|
>HA axis may be overriden by higher centres of the brain
- e.g. in chronic stress, HPA becomes hyperactive with loss of negative feedback to CRH and ACTH, and other elements of anterior pituitary (e.g. gonadotrophs and somatotrophs) repressed following withdrawal of hypothalamic releasing hormones |
|
|
|
How may communication between the hypothalamus and the pituitary be disrupted?
|
1. Cranial trauma
2. Pressure on the pituitary stalk usually secondary to a pituitary tumour (occlusion of hypophyseal portal circulation and or supraoptic hypothalamic tract 3. Inflammation / infection 4. Congenital defects of midline structures (septo-optic dysplasia = panhypopituitarism with blindness) 5. Kallman's syndrome (absence of GnRH neurones from the hypothalamus |
|
|
|
Anterior pituitary cells are sensitive to irradiation, which cells are most sensitive?
|
>Somatotrophs - lose function progressively
- isolated deficiencies of single adenohypophyseal hormones are rare |
|
|
|
What forms of pituitary tumour are most common?
|
FUNCTIONAL TUMOURS ARE RARE:
>Prolactinoma = most common - can be shrunk by dopamine agonists e.g. bromocriptine - cause galactorrhea and suppression of the HPG axis >Adenocorticotrophic tumours - Cushings disease - caused by hypersecretion of ACTH from anterior pituitary tumours - tend to be hyperpigmented, classically with acanthosis nigricans |
|
|
|
Why is ACTH secreted from small cell tumours of lung?
|
Secondary to adenocarcinoma of pituitary
|
|
|
|
TSH secreting tumours are very rare - what do elevated levels of TSH usually signal?
|
>Underactive thyroid gland or assay errors
|
|
|
|
What are the functions of Oxytocin?
|
>Oxytocin elevates intracellular Ca++ to stimulation contraction of smooth muscles of
- breast (to stimulate milk ejection) - uterus (to stimulate expulsion of infant at parturition) |
|
|
|
What triggers the secretion of OT from the posterior pituitary?
|
>Secretion of OT from posterior pituitary triggered by neural reflex inputs
- suckling reflex (plus sensory stimulus of crying infant) - Fergusson reflex (isometric contractions of myometrium plus distension of vagina) |
|
|
|
What may be administered to assist in parturition?
|
>Oxytocin analogues e.g. syntocin
|
|
|
|
What is gestational diabetes?
|
>Severe insulin resistance caused by hormonal changes and metabolic stress of pregnancy
>In women with low insulin secretory capacity this can result in a form of diabetes >Affects 2% of pregnancies of non diabetic women >Symptoms normally disappear after birth but 60% of these women go onto develop diabetes later in life |
|
|
|
What occurs in hypersecretion of GH from the pituitary?
|
>Acromegaly and gigantism
|
|
|
|
What type of patient would be expected in a thyroid clinic?
|
>Predominantly female
- F:M ratio about 10:1 for the major thyroid diseases >Postmenopausal age - thyroid disease incidence increases with age - childhood problems are rare with some important exceptions |
|
|
|
How are thyroid clinic patients typically referred?
|
>Referred by their GP
- often grumble how long it took - insidious onset >Often initial symptoms attributed to other causes e.g. old age, stress, 'late nights' etc. - can see the marked effects of simple treatment |
|
|
|
Do thyroid patients look 'at home' in the clinic?
|
>Yes
- many attending periodic follow up appointments e.g. annual checkup - only a few new patients per clinic |
|
|
|
What symptoms might a thyroid clinic patient complain of?
|
2 main types
- overweight / lethargic / unengaged - thin / hyperactive / fidgeting / exopthalmos >new patients may be particularly worried - feel lumps in their neck - need reassurance that thyroid nodules are common due to the follicular structure of the thyroid - thyroid cancer rare |
|
|
|
Are there any obvious thyroid clinic symptoms?
|
>A few may show gross symptoms i.e. eye signs or goitre
- however most show signs only evident to the trained eye |
|
|
|
Which specialist publications might be available to a thyroid clinic patient?
|
>Newsletters from patient support groups
British thyroid foundation (7000 members) - Patient support and information - Network regional groups - Awareness amongst the public and medics - Raise funds for research Thyroid eye disease newsletter (800 members) - smaller specialist group - for unfortunate patients with thyroid eye disease - youngest member only 13 years old, suffered severe bullying at school |
|
|
|
Who typically directs the thyroid clinic?
|
>Specialist thyroid consultant (likely a member of the British Thyroid Association)
- physician - endocrinologist - diabetologist >Close relationship with 3 other specialists - chemical pathology - nuclear medicine / imaging - thyroid surgeon |
|
|
|
What questions are thyroid clinic patients likely to be asked in a consulting room environment?
|
- appetite
- weight - energy levels - ability to cope with stress - general mood - depression? - sleeping patterns - temperature regulation - general skin problems (more an issue for hypothyroid) - localised skin problems - eyes - palpitations - menstrual irregularity - bowel movements - libido - family history |
|
|
|
What physical examination might a thyroid patient be given?
|
- palpate thyroid gland for nodules
- examine eyes - check cardiac well-being - reflexes - tremor - acropachy - dry / sweaty skin and hair - pretibial myxodema |
|
|
|
Who else might a thyroid clinic patient be referred to for further investigation?
|
1. Chemical pathology
- blood sample - TFTs - Measure TSH, T4 and T3 by immunoassay 2. Immunology - blood sample - try to detect autoantibodies against the thyroid 3. Imaging e.g. nuclear medicine - radioisotope or ultrasound - looking for hot / cold nodules 4. Histology - fine needle aspiration - check details of follicular structure |
|
|
|
What are the most frequently encountered clinical issues in the thyroid clinic?
|
1. Hyperthyroidism
- overactive gland - 50% due to Graves' disease - 50% autonomous nodules 2. Hypothyroidism - underactive thyroid gland - possibly Hashimoto's thyroiditis |
|
|
|
What treatments are offered to thyroid patients?
|
Well established treatments
- T4 replacement therapy - thyroid surgery (therapeutic or cosmetic) - antithyroid drugs: inhibit T3/T4 synthesis - radioactive iodine More rarely - immunosuppression - orbital decompression |
|
|
|
Are there cures for thyroid issues?
|
>Pathology well understood
- frequent autoimmune origin - can manage symptoms well - not a cure >Thyroid eye disease - pathology still not understood - fortunately rare - can be difficult to treat |
|
|
|
Are there side effects to thyroid medications?
|
Not usually - most thyroid patients can be managed very successfully given time
|
|
|
|
Will thyroid patients be on treatment for long?
|
Wide range of possibilities
- on T4 replacement for life (daily pill) - on antithyroid drugs for a few months - radioactive iodine: ideally just one treatment, patient may be isolated for a few hours / days afterwards - immunosuppression for one month |
|
|
|
How often will thyroid patients be required to attend the clinic in the future?
|
>Initially quite frequently on an outpatient basis
- e.g. every few weeks - results from tests / diagnosis - initial treatment >Long term management can be provided by GP with maybe periodic e.g. annual checks by the thyroid clinic |
|
|
|
How well is the thyroid clinic established?
|
Yes! Especially at UCL
>Harrington and Pitt Rivers - structure of T4 >Pochin - Radioiodine used for first time in UK >Doniach - discovered thyroid autoimmunity >Pitt Rivers - Discovered T3 >Ekins - Discovered immunoassay and used for T4 |
|
|
|
What embryologyical abnormalities of the thyroid occur?
|
>Failure of the gland to develop - congenital hypothyroidism
>Under or over migration of the thyroid - lingual or retrosternal thyroid >Failure of thyroglossal duct to atrophy - thyroglossal cyst >Congenital absence of the thyroid due to mutation in genes such as PAX8 |
|
|
|
What is the morphology of the thyroid gland?
|
>2 lobes
>15-40g >Situated below the larynx >Normally quite difficult to see and feel >When grossly enlarged = goitre >4 parathyroid glands attached to posterior surface - synthesise PTH |
|
|
|
What is the microscopic morphology of the thyroid gland?
|
|
THYROID MORPHOLOGY
|
|
|
How do follicles differ in i) underactive thyroid vs. ii) overactive thyroid?
|
>Underactive follicles with flattened thyroid epithelial cells and increased colloid
>Overactive follicles with tall columnar epithelial cells and reduced colloid |
|
|
|
What occurs during thyroid hormone biosynthesis?
|
>Thyroid hormones is carried out by the enzyme thyroid peroxidase, an integral membrane protein present in the apical (colloid facing) plasma membrane of thyroid epithelial cells
>Thyroid peroxidase catalyses two sequential reactions - iodination of tyrosines on thyroglobulin (organification of iodide) - synthesis of thyroxine or triiodothyronine from two iodotyrosines >Through the action of thyroid peroxidase, thyroid hormones accumulate in colloid on the surface of thyroid epithelial cells - hormone is still tied up in molecules of thyroglobulin - the task remaining is to liberate it from the scaffoled and secrete free hormone into blood |
THYROID HORMONE BIOSYNTHESIS
|
|
|
How is thyroid hormone released from the thyroid follicles?
|
>Released from their thyroglobulin scaffold by digestion in lysosomes of thyroid epithelial cells
- epithelial cells ingest colloid by endocytosis from their apical borders, which contains thyroglobulin decorated with thyroid hormone - colloid laden endosomes fuse with lysosomes which contain hydrolytic enzymes that digest thyroglobulin, thereby liberating free thyroid hormones - free thyroid hormones apparently diffuse out of lysosomes through the basal plasma membrane of the cell and into blood where they quickly bind to carrier proteins for transport to target cells |
|
|
|
What are the clinical uses of the iodide pump?
|
1. Small dose: image gland with a gamma camera
2. Single high dose - ablates overactive thyroid - given to hoover up secondaries in treatment for thyroid cancer after thyroid removal by surgery |
|
|
|
In what two ways might tyrosine residues be iodinated on thyroglobulin?
|
1. +I- = monoiodotyrosine
2. +2I- = diiodotyrosine |
|
|
|
How is T3 formed differently from T4 within the TG?
|
T3=MIT + DIT
T4=DIT + DIT |
|
|
|
How many months store of iodinated Tg is present in the thyroid?
|
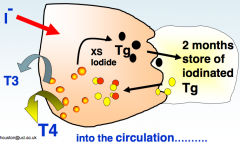
|
T3 T4 SYNTHESIS
|
|
|
What are the normal ranges for thyroid hormones?
|
NORMAL TOTAL CONCNS:
>T4 = 60-150 nM >T3 = 1.2-2.9 nM |
|
|
|
How is T3/T4 transported in the blood?
|
>99% bound to 3 different proteins
- thyroxine binding protein - binds both T4 and T3 despite name |
|
|
|
What is the rate of blood flow in the thyroid?
|
4-6 mls / min / g tissue
- x2 that of kidney - High rate important for delivery of I- and TSH - secretion of T3 and T4 >Responsible for the audible bruit of the overactive gland |
|
|
|
What are the physiological actions of T3 and T4?
|
Generalised effects:
1. Increases BMR 2. Promotes balanced growth and development 3. Main actions - modulates the actions of other hormones e.g. Insulin, adrenaline, TRH, GHRH |
|
|
|
How does T4 differ from T3 aside from its chemical structure?
|
>T4 is regarded as a prohormone for T3
- in target cells, a deiodinase immediately converts T4 to T3 - T3 acts on nuclear receptors, target cell decides how much T3 it needs, it isn't a passive victim to T4 levels in the circulation :. T4, forms a huge circulating store for T3 |
|
|
|
What is rT3?
|
Target cells have the ability to convert T4 into reverse T3 which has no known biological activity
- potentially a mechanism for the target cell to get rid of excess T4 |
|
|
|
Where does T3 act?
|
>On nuclear receptors
- member of steroid thyroid hormone receptor superfamily - bound to DNA in their resting state >Occupancy -> transcriptional activation - often dimerise with receptors for retinoic acid |
|
|
|
How should these results be interpreted?
|
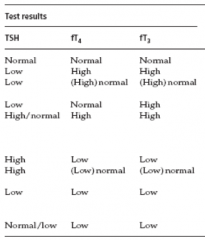
TFT QUESTIONS
|
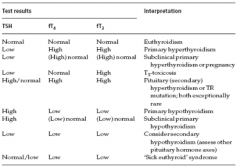
TFTS
|
|
|
Patient with:
T3 = 4.6nM/L T4 = 210 nM/L TSH = 0.1mU/L What is your diagnosis? |
Primary hyperthyroidism, possibly Graves' disease.
|
|
|
|
Patient with:
T3 = 1.2nM/L T4 = 40 nM/L TSH = 10mU/L What is your diagnosis? |
Primary hypothyroidism, possibly Hashimoto's Thyroiditis
|
|
|
|
Patient with:
TSH = 20mU/L What is your diagnosis? |
>Primary Hypothyroidism, possibly Secondary Hyperthyroidism but extremely rare, so unlikely
|
|
|
|
Patient with:
TSH = 0.2mU/L What tests do you request to make a diagnosis? |
T4 and T3 levels, likely a primary hyperthyroidism
|
|
|
|
Patient with:
T3 = 0.8nM/L T4 = 35nM/L TSH = 25mU/L, patient returns from 2 week trek in Nepal. What is your diagnosis? |
>Primary hypothyroidism, 2 weeks in Nepal irrelevant due to presence of a 2 month store of iodinated thyroid hormones present in thyroid, in addition to an ability to stretch euthyroid characteristics a further month once existing stores are depleted
|
|
|
|
How is thyroid function measured and why?
|
>T3/T4 negative feedback on TSH production is the basis of TFTs
>Excess unregulated production of TSH from the pituitary thyrotrophs e.g. by a tumour is exceedingly rare - therefore TRH not routinely measured |
|
|
|
What is meant by pituitary 'lag'?
|
>in a hypothyroid patient it takes a number of days for T4 replacement therapy to lower TSH
- can be used to check compliance |
|
|
|
What forms of goitre are there?
|
>Smooth
>Multinodular >Isolated nodule |
|
|
|
What is goitre associated with?
|
1. Iodine deficiency
2. Hypothyroidism 3. Hyperthyroidism |
|
|
|
What is the prevalence of hyperthyroidism (UK)?
|
>4% in the UK
|
|
|
|
What are the features of hyperthyroidism?
|
>Gives rise to thyrotoxicosis
>Biochemical features - usually both T3 and T4 are raised - suppressed TSH (-ve feedback) >However 5% are T3 toxic - only T3 raised, not T4 - suppressed TSH >Virtually never due to raised TSH |
|
|
|
What are the causes of hyperthyroidism?
|
>Graves' disease
>Toxic multinodular goitre >Solitary toxic adenoma >Thyroiditis >Exogenous iodine and iodine containing drugs e.g. amiodarone >Excessive T4 or T3 ingestion >Ectopic thyroid tissue e.g. struma ovarii >Functioning metastatic thyroid cancer |
|
|
|
What are the clinical features of hyperthyroidism (general to thyrotoxicosis)?
|
>Weight loss, normal appetite
>Sweating and heat intolerance >Agitation >Tremor >Fatigue >Palpitation / atrial fibrillation >Angina >Generalised muscle weakness (proximal myopathy) >Diarrhoea >Oligomenorrhoea and infertility >Goitre >Eyelid retraction and lig lag |
|
|
|
What are the features of hyperthyroidism peculiar to Graves' disease?
|
>Periorbital oedema
>Proptosis >Diplopia >Corneal ulceration >Loss of visual acuity >Pretibial myxedema >Thyroid acropachy |
|
|
|
Which disease causes 50% of thyrotoxicosis?
|
>Graves' disease
- autoimmune disease - autoantibody against the TSH receptor - binds and activates the receptor - only well established example of an antibody which mimics a hormone |
|
|
|
What are the treatments for hyperthyroidism?
|
1. Anti thyroid drugs
- e.g. carbimazole - inhibit T3 and T4 biosynthesis by the thyroid 2. Thyroid ablation - gamma irradiation with 131 Iodide - patient drinks a solution of sodium 131 iodide 3. Thyroid surgery - partial or complete removal of thyroid (thyroidectomy) - implantation of parathyroids |
|
|
|
What is the typical treatment strategy for Graves' disease? Why?
|
>Graves' disease is autoimmune, therefore may spontaneously remit
- initially patient is rendered euthyroid with CBZ - continued for 9-12 months - withdraw CBZ, observe patient over a few weeks >If patient remains euthyroid then left alone >If patient relapses - offer thyroid ablation or surgery |
|
|
|
What is the treatment for non-Graves' thyrotoxicity?
|
>Since there is no chance of a spontaneous remission
- render euthyroid with CBZ - offer thyroid removal, ablation, surgery |
|
|
|
What are the factors to consider when offering radioablation or surgery?
|
>Personal factors must be considered
- age and sex - 131 Iodide offered to adult men and postmenopausal women - surgery offered to young people (to avoid rendering them infertile) |
|
|
|
What are the adverse effects of CBZ?
|
>Agranulocytosis
>Thrombocytopenia >Aplastic anaemia |
|
|
|
What are the adverse effects of radioiodine / surgery?
|
>May render patient hypothyroid
- requiring T4 for life >Surgery may damage the recurrent laryngeal nerve |
|
|
|
What is the prevalence of hypothyroidism in the UK?
|
2%
|
|
|
|
What are the characteristics of hypothyroidism (myxodema)?
|
>Often autoimmune in origin (Hashimoto's disease)
>Not to be confused with pretibial myxedema >Biochemical features: PRIMARY - T4 low / TSH raised SECONDARY - due to pituitary failure - T4 low, TSH suppressed |
|
|
|
What is produced in Hashimoto's disease?
|
>Antithyroglobulin Abs
>Anti peroxidase Abs |
|
|
|
What are the causes of hypothyroidism?
|
>Autoimmune (Hashimoto's)
>Post surgery or radioiodine >Congenital >Dyshormogenesis >Secondary to pituitary or hypothalamic disease >Iodine deficiency |
|
|
|
What are the clinical features of hypothyroidism?
|
>Lethargy, tiredness
>Cold intolerance >Dry / coarse hair >Hoarseness >Weight gain >Slow muscle and tendon reflexes >Anaemia >Dementia >Constipation >Bradycardia >Infertility >Galactorrhea |
|
|
|
What are the treatments for hypothyroidism?
|
>Hormone replacement therapy
- daily T4 tablets - emergency e.g. myoedematous coma T3 |
|
|
|
What is the prevalence of of hypothyroid neonates?
|
>1:4000 live births in an iodine replete region
>Due to congenital hypothyroidism >Danger of cretinism - severe neurological impairment - all new borns are screened for raised TSH (heel prick) - treated with T4 asap |
|
|
|
What is our iodide requirement (daily)?
|
50ug
|
|
|
|
What are the sources of iodide for humans?
|
>Main include
- drinking water, fish, milk - supply depends on local geology - problem typically in mountainous central land masses, e.g. European and the Southern Alps in NZ, Himalayas |
|
|
|
To what degree can the thyroid compensate for iodine insufficiency?
|
>Through increased iodide uptake and scavenging
>Preferential formation of MIT over DIT - increasing T3 : T4 >Maintains a euthyroid status for a while |
|
|
|
What is the treatment for iodine insufficiency?
|
>Dietary supplementation
- salt - bread >Iodised oil (depot injections) |
|
|
|
Name some different examples of growth deficiency disorders?
|
1. Achondroplasia
2. Malabsorption syndrome 3. Classic growth hormone deficiency 4. Idiopathic growth hormone deficiency 5. Laron dwarfism 6. Hypothyroidism 7. Juvenile Cushing's 8. Pseudohypoparathyroidism 9. Turner's syndrome 10. Delayed puberty 11. Anorexia nervosa 12. Intrauterine growth retardation 13. Cranial irradiation 14. Chronic illness 15. Psychosocial deprivation |
|
|
|
Name some different examples of excessive growth disorders?
|
1. Gigantism
2. Acromegaly |
|
|
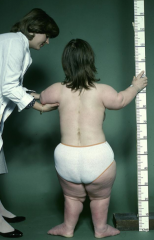
What is this an example of? What are the signs?
|
ACHONDROPLASIA
>Disproportional growth failure >Short limbs >Normal GH >No response to GH" |
ACHONDROPLASIA
|
|
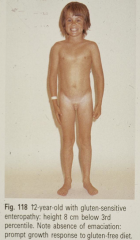
What is this an example of? What are the signs?
|
MALABSORPTION SYNDROME - COELIAC DISEASE
- Proportional growth faiiure - GH normal - Prompt response to gluten free diet |
MALABSORPTION SYNDROME
|
|
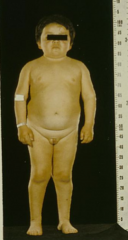
What is this an example of? What are the signs?
|
CLASSIC GH DEFICIENCY
- proportional growth failure - immature face - truncal obesity - little or no GH - 1:4000 prevalence - usually idiopathic - hypothalamic disorder? |
CLASSIC GROWTH HORMONE DEFICIENCY
|
|

What is this an example of? What are the signs?
|
IDIOPATHIC GH DEFICIENCY
- twins - unknown cause - proportional short stature |
IDIOPATHIC GROWTH HORMONE DEFICIENCY
|
|

What is this an example of? What are the signs?
|
LARON DWARFISM
- excessively short - immature face - proportionate limb growth - GH normal or raised |
LARON DWARFISM
|
|
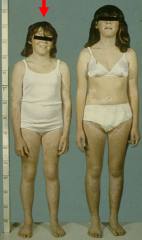
What is this an example of? What are the signs?
|
HYPOTHYROIDISM
- short limbs - not achieving full growth potential - GH low - T3/T4 low |
HYPOTHYROIDISM
|
|
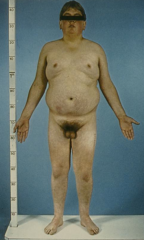
What is this an example of? What are the signs?
|
JUVENILE CUSHING'S SYNDROME
- proportionate limbs - not achieving full growth potential - truncal obesity - symptoms of excess of cortisol - GH low |
JUVENILE CUSHINGS
|
|
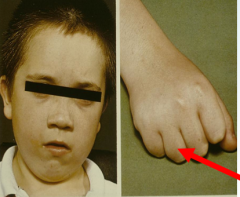
What is this an example of? What are the signs?
|
PSEUDOHYPOPARATHYROIDISM
- proportionate limbs - not achieving full growth potential - round face - short 4th metacarpal - high PTH but resistance to PTH - normal GH |
PSEUDOHYPOPARATHYROIDISM
|
|
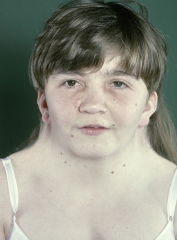
What is this an example of? What are the signs?
|
TURNER'S SYNDROME
- XO - short stature - webbed neck |
TURNER'S SYNDROME
|
|
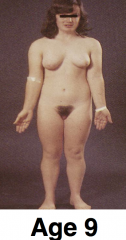
What is this an example of? What are the signs?
|
PRECOCIOUS PUBERTY
- early sexual maturity - initial rapid growth - but growth stops early - premature epiphyseal fusion |
PRECOCIOUS PUBERTY
|
|
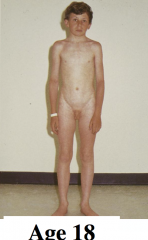
What is this an example of? What are the signs?
|
DELAYED PUBERTY
- puberty after 15 years - 1:50 prevalence - M:F ratio is 6:1 - growth slows around year 10 - height falls below 3rd centile - bone age delayed - LH/FSH - GH normal catch up growth occurs - reaches full growth potential |
DELAYED PUBERTY
|
|
|
What are the characteristics of anorexia nervosa?
|
>Prevalence 1:100
>Hypothalamic turn-off >Low GnRH release >Fasting GH and cortisol |
|
|
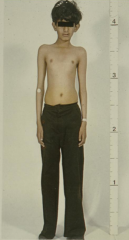
What is this an example of? What are the signs?
|
"INTRAUTERINE GROWTH RETARDATION
- low birth weight - placental insufficiency - poor catch-up growth " |
INTRAUTERINE GROWTH RETARDATION
|
|

What is this an example of? What are the signs?
|
GIGANTISM
- high pre-pubertal GH - proportional limb growth - surpasses growth potential |
GIGANTISM
|
|

What is this an example of? What are the signs?
|
ACROMEGALY
- high GH post puberty - no increase in linear growth - increased foot and hand size - coarsening of facial features, prominent frontal ridge and prognathism |
ACROMEGALY
|
|
|
What are the important considerations when diagnosing growth abnormalities?
|
>Highly diverse clinical conditions are associated with short stature
- :. we need very careful history taking and many special features to be considered >Key clues ought to be considered - balanced limb growth - chondrocytes are important for growth of long bones - adequate nutrition is required - stress inhibits growth - anterior pituitary / somatotrophs / GH vital / prepuberty - low GH = short stature - high GH = gigantism - window of time - prepuberty - sex steroids also play a role - other hormones e.g. PTH, T3/T4, cortisol modulate GH production and influence - genetic factors set growth potential >Balanced growth depends upon a blanaced environment and a balanced endocrine system |
|
|
|
Why is short stature a problem?
|
>As children / adolescents it causes bullying etc.
>As adults it is a disadvantage in our competitive society |
|
|
|
What anthropometric measurements are used to assess growth?
|
1. Height v. chronological age (individual)
2. Rate of growth (velocity) v. chronological age 3. Bone age / bone maturation 4. Pubertal stages 5. Height v. chronological age (population) 6. Standard deviation score |
|
|
|
How is height v. chronological age used to assess patients?
|
<2 years: supine length
>2 years: standing height - stadiometer used - carefully position individual - check technique (claim 1% CV attainable) - repeat at regular time intervals |
|
|
|
How is rate of growth v. chronological age used to assess patients?
|
>Smooths out slightly erratic single height measurements
>Very high in first year of life >Plateau followed by pubertal growth spurt >Sharp decline at end of puberty to zero |
|
|
|
How is bone age used to assess patients?
|
>Assessed from radiographs of the wrist
>Compare epiphyseal growth plates against age-related reference images >Cumulative scores for several bones: claim 2% CV attainable >Difficult for children with skeletal dysplasias |
|
|
|
How are pubertal stages used to assess patients?
|
>Important for assessing delayed or precocious puberty
- compare stages of pubic (6, boys and girls) and axillary (3, boys) hair growth against reference photographs >Boys: 5 developmental stages of external genitalia assessed using reference photographs and also an orchidometer for testicular size >Girls: 5 breast development stages assessed against reference photographs |
|
|
|
How is height v. chronological age of the population used to assess patients?
|
>Height percentiles used
NB. - appropriate reference population must be used - generations change - linked to improved nutrition and economic conditions |
|
|
|
How is a standard deviation score used to assess patients?
|
= (observed height - mean height for age and sex)/SD for that age and sex
- normal values lie from -2 to +2 |
|
|
|
What is the dominant endocrine regulator of growth?
|
>Growth hormone
- however for the first year growth is largely dependent on nutrition |
|
|
|
When do GH receptors appear in humans?
|
>at 7 months
|
|
|
|
Where is growth hormone secreted from, how is it stored?
|
>Secreted from somatotrophs in the anterior pituitary
>Preformed granules - 10% pituitary dry weight - 10mg / pituitary |
|
|
|
What is the structure and half life of growth hormone?
|
>Single chain protein
- 2 disulphide bonds - 2 forms (22kD, 20kD) >T1/2 = 15 minutes >Similar in structure to prolactin |
|
|
|
What is the structure of the GH receptor?
|
Single transmembrane receptor:
- structure is related to leptin - cytokines e.g. interleukins and EPO - 4 long antiparallel alpha helices |
|
|
|
Does non human GH work in man?
|
>No e.g. differs from bGH by 35%
- however hGH can act on lower mammalian orders e.g. cattle |
|
|
|
What are the members of the human GH gene family?
|
>Located on chromosome 17
- GH and CS (chorionic somatomammotropin) >GH-N is expressed preferentially in the somatotropes of the anterior pituitary, while GH-V and the CS genes are expressed in the syncytiotrophoblasts of the placenta >HS I and II are pituitary specific >HS IV is placenta specific >III and V are found in both the pituitary and placenta but nowhere else |
|
|
|
Why is the species specificity of GH important clinically?
|
>Human GH must be used in GH deficient children
>Pre-1985, hGH was extracted from human pituitaries - risk of Creutzfeld Jakob disease - now it is produced recombinantly |
|
|
|
Which 2 receptor groups respond to GH?
|
>Lactogenic receptors: both GH and PRL activate
>Somatogenic receptors: only GH activates |
|
|
|
What is the functional morphology of the GH receptor?
|
>Cell surface receptors with three domains
- extracellular / transmembrane / cytoplasmic >GH + two GH receptors form a complex - sequential dimerisation occurs which activates intracellular signalling via the JAK/STAT pathway and MAPK and PI3 pathways - the extracellular domain is shed, which also functions as a circulating GH binding protein |
|
|
|
What are the physiological actions of GH?
|
1. Metabolic
- decreases glucose metabolism - opposing insulin - increases lipolysis - increases protein synthesis (anabolic) - increases milk yield in cows 2. Other - increases IGF production from liver etc - increases chondrocyte and cartilage formation 3. Growth promotion - clonal expansion of chondrocytes - growth of bones, soft tissue and viscera |
|
|
|
What is the structure and function of IGF-1?
|
>Similar to proinsulin + red tail
- IGF-1, but not IGF-2 acts via insulin like receptors - also known as somatomedin C >Transforms fibroblasts - into osteoblasts - into chondrocytes - into fat cells |
|
|
|
What is the cause of achondroplasia?
|
>Defective chondrocyte growth in growth plates
>Leads to short long bones and limbs >Due to defective receptors for fibroblast growth factor (FGF) >Therefore not responsive to growth hormones |
|
|
|
What marker is used as an in vivo bioassay for GH?
|
>Increase in growth plate thickness
- remove pituitary from prepubertal rat - inject GH - remove tibia / cut bone sections - measure growth plate thickness |
|
|
|
What occurs at the end of puberty which causes growth velocity to decline and halt?
|
>Sex steroids have increased
- androgens in boys - oestrogens in girls >These cause the growth plates to close, stopping growth NB. AROMATASE has been found in growth plates in boys and girls - converts testosterone into oestradiol - thus oestradiol closes growth plates in both boys and girls - aromatase inhibitors can be used to delay plate closure in boyes but not for girls |
|
|
|
What are the physiological actions of GH?
|
>Wide spectrum, multiple target tissues
>Most notable increased bone growth and weight >Direct metabolic actions tend to oppose insulin >Indirect metabolic actions via IGFs tend to be insulin like |
|
|
|
How is GH secretion regulated?
|
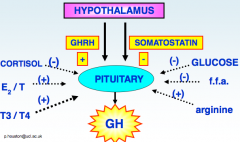
>Both GHRH (10aa) and somatostatin (14aa) are small hypothalamic peptides
- Both act on G-protein coupled receptors on somatotrophs: - GHRH activates adenylate cyclase - Somatostatin acts via PLC >Androgens & oestrogens sensitize somatotrophs to GHRH - This accounts for the pubertal rise in GH and growth spurt - Metabolic products influence GH release – used for clinical tests - aa such as arginine stimulate GH release - glucose and free fatty acids suppress GH release |
REGULATION OF GH
|
|
|
How is GH measured?
|
IMMUNOASSAY
- ELISA (Enzyme Linked Immunosorbent Assay, since GH is a 22kDa protein - large-scale routine use - very precise and sensitive - does not measure biological potency directly BIOASSAYS - Used to measure potency of new batches of recombinant GH - Rarely used to check bioactivity of patient GH - In vivo/ hypophysectomised rats/ measure increase in growth plate thickness |
|
|
|
Which tools are used in the diagnosis and management of growth disorders?
|
1. Clinical observation
- history rule out multiple non-endocrine causes e.g. respiratory, cardiact, renal problems - proportionate growth in appearance? - anthropometric measurements: growth velocity, bone age, pubertal stages etc. 2. Measure growth hormone by immunoassay - special points to note re interpretation of serum GH values i) pulsatile secretion pattern - 24hr profile - sampling interval 20 minutes - major peaks during stages 3 or 4 - delayed sleep = delayed rise in GH - difficulties for interpretation / random samples of limited value - 24hr profile inconvenient for the patient and lots of work for the lab ii) abnormalities Classic idiopathic GH deficiency - absent or only feeble peaks GH excess - continuous secretion of high level - absent pulsatility iii) dynamic function tests for GH secretion - widely used, deal with the problem of irregular secretion iv) GH secretion naturally declines with age - during and after the 3rd decade |
|
|
|
What two dynamic function tests may be used to test GH secretion?
|
1. Provocative tests
- to test pituitary reserves - inject insulin: sample every 20 minutes for 4 hours and measure GH >This is known as an insulin tolerance test - risk of hypoglycaemia - alternative stimulators include GHRH, Bovril and exercise - rise in GH is due to the fall in glucose rather than the insulin itself >A normal response is a bell shaped secretion of serum GH over the four hours rising to a peak of 100mU/L 2. Suppression tests - drink glucose solution: sample every 20 minutes for 4 hours and measure GH >The normal response should show a depression in GH release which increases towards the end of the four hours - no suppression seen for excess GH conditions e.g. pituitary tumour - suppression occurs to to a rise in glucose - late rise due to the fall in glucose in response to the natural rise in insulin (i.e. you have a mini ITT naturally induced at the end of the suppression test) |
|
|
|
Which other three proteins may be measured to test GH?
|
1. IGF levels
- also measured by immunoassay - not as widely used as GH measurements (confined to specialist centres 2. IGF binding proteins - measured in special centres for difficult cases - still research rather than routine 3. GH binding proteins - measured in the highly specialised centres managing the rare cases of Laron Dwarfism - GHBP = ectodomain of the GH receptor therefore provides a convenient check on receptor integrity - measurement methods not well established |
|
|
|
What are the key diagnosis points for idiopathic GH deficiency?
|
- proportionate growth and relatively normal appearance
- truncal obesity and immature face - low rate of growth: low SDS score - nil or poor GH response to an ITT provocation test - nil or feeble peaks in a 24hr profile - low IGF-1 |
|
|
|
How is idiopathic GH treated?
|
>Depends on whether a sufficient prepubertal window of time is available
- daily subcut injection - usually at night - expensive - different countries have different cut off criteria |
|
|
|
Which patients with short stature are growth hormone resistant? Why?
|
LARON DWARFS
- first described by Zvi Laron in 1966 - Good example of a classic hormone resistant condition >Key points for diagnosis - rare, autosomal recessive condition - very low SDS score and growth velocity - proportionate but with very immature face - normal GH levels - low IGF-1 >most severe forms: no binding to circulating GHBP >less severe forms: reduced binding affinity - there may well be a spectrum of severity depending on the mutation site - in the most severe formes large deletions in the GH receptor have been identified |
|
|
|
How is Laron dwarfism treated?
|
>hGH not effective
>Trials with recombinant IGF-1 in progress |
|
|
|
What occurs with growth hormone secretion in gigantism (before puberty)?
|
BEFORE PUBERTY
>Due to a benign tumour of somatotrophs - autonomous, unregulated, non pulsatile secretion - incidence of tumour = PRL>>ACTH>GH, those secreting LH/FSH/TSH very rare >Continuous high prepubertal GH levels distort growth causing - excessive early growth - delayed puberty (due to compromised pituitary function and low LH/FSH, somatotroph compression of other cells in the restricted space of the sella turcica) - Prolonged pre-pubertal linear growth period prior to an exaggerated growth spurt - results in excessive height attainment - poor prognosis if untreated |
|
|
|
What are the various prognoses for gigantism?
|
>Easily recognised syndrome
- nowadays a high Z score should be picked up early during childhood and the tumour which can be imaged with an MRI can be removed surgically (transsphenoidal adenomectomy) >However - in some cases impaired endocrine function can recover, when the normal but compressed pituitary has been preserved - for others, difficulties in growth, development and sexual maturation may persist, however with careful medical management with hormone replacement therapy a normal quality of life can be expected |
|
|
|
What are the available treatments for GH excess?
|
Medical
- dopamine agonists (inhibit GH release) e.g. bromocriptine - octeotride - somatostatin analogue - pegvisomat: mutated GH - prevents receptor dimerisation Surgical - transphenoidal route usual - guided by MRI scans - transfrontal route used if the tumour is large Radiotherapy - conventional supervoltage - slow effect in reducing tumour secretion of hormones and tumour shrinkage, usually results in hypothyroidism many years later |
|
|
|
What is the usual lifetime pattern of GH secretion?
|
>Foetus: growth largely stimulated by IGF-2
>Infancy: malnutrition rather than GH/IGF-1 very important in first year >Postnatal growth is rapid e.g. 15cm per year >Declines at 3 years to a plateau (about 6cm/year) >Followed by pubertal rises in GH/IGF-1 and the growth spurt >Estrogens close growth plates at end of puberty >Thereafter GH steadily declines throughout adult life - the somatopause, central adiposity, decreased muscle mass |
|
|
|
What are the advantages of immunoassays as a diagnostic test in medicine?
|
1. Very SENSITIVE
- can detect picomolar concentrations - due to the high affinities of antibodies for antigens - need only small quantities of serum e.g. 10uL 2. Highly hormonally SPECIFIC - can distinguish between T3 and T4 and between different steroids - due to the high specificity of antigen recognition 3. Very PRECISE - simple steps with micropipettes and responses measured with instruments capable of highly reproducible signal measurements e.g. radioactivity counters, spectrophotometers 4. High sample THROUGHPUTS - simple steps with specially designed equipment e.g. microtitre plates for ELISAs , with 96 optical densities being read in 20 seconds 5. CHEAP - inexpensive reagents used at high dilutions - very profitable to run for e.g. a private pathology service 6. Highly developed QUALITY CONTROL systems in e.g. the UK - nationwide basis (NEQAS) - monthly checks on all immunoassay labs - T4 measured across the UK with as little as 10% variance across the country |
|
|
|
What occurs with growth hormone secretion in gigantism (after puberty)?
|
>No increased height is now possible because of oestrogen closing the growth plate
>Annual incidence of acromegaly about 3 per million >Insidious onset - in post mortem series about 25% of the population has evidence for a pituitary tumour >Raised GH causes raised circulating IGF-1 - proliferation of bone, cartilage and soft tissues and increase in size of other organs - enlarged hands and feet, jaw protrusion and dental malocclusion >Raised GH opposes insulin - risk of diabetes mellitus with associated symptoms - e.g. polydipsia, polyuria, recurrent UTIs, retinopathy, neuropathy |
|
|
|
A normal adult body contains how much calcium?
|
>1 kg (25mol) calcium
|
|
|
|
How much calcium is present in bone, and in what form?
|
>99% as hydroxapatite
- remaining 1% outside the skeleton in ICF and ECF and tissues |
|
|
|
Growing infants are in positive calcium balance, how much does their calcium content typically increase by?
|
0.4g (10mmol) per day
|
|
|
|
How much calcium must be absorbed by the body each day to balance the daily losses of calcium?
|
1g or 25mmol
|
|
|
|
What percentage of filtered calcium is typically reabsorbed in the kidneys?
|
98%
|
|
|
|
What are the normal plasma concentrations of calcium?
|
>2.2-2.6mmol/L
|
|
|
|
Where is calcium reabsorbed in the kidney? How is it regulated?
|
PROXIMAL TUBULE
- paracellular transport, not regulated by hormone THICK ASCENDING LIMB - paracellular transport, not regulated by hormones - transcellular transport - increased by parathyroid hormone - reduced by calcitonin DISTAL TUBULE - transcellular transport - increased by calcitriol and parathyroid hormone - decreased by calcitonin |
|
|
|
What are the normal plasma concentrations of phosphate?
|
>0.6-1.3
|
|
|
|
What proportion of plasma calcium exists associated with albumen, citrate and uncomplexed?
|
>Albumen = 44%
>Citrate = 9% >Ca++ ions = 47% |
|
|
|
Name two cellular roles for calcium.
|
1. Cardiac action potentials
2. Muscle fibre contraction |
|
|
|
In which tissue and cell type is PTH produced?
|
>Parathyroid glands - chief cells
|
|
|
|
In which tissue and cell type is calcitriol produced?
|
>Skin - caliciol
>Liver - calcidiol >Kidney - calcitriol |
|
|
|
In which tissue and cell type is calcitonin produced?
|
>Thyroid gland - C cells
|
|
|
|
What cells are APUD cells?
|
>Cells which have the following characteristic:
- Amine - for high amine content. - Precursor Uptake - for high uptake of (amine) precursors. - Decarboxylase - for high content of the enzyme amino acid decarboxylase (for conversion of precursors to amines) |
|
|
|
Which APUD cells are derived from neural crest?
|
e.g. Chromaffin cells of the adrenal medulla
|
|
|
|
Which APUD cells are not derived from the neural crest?
|
>Islets of Langerhans
>Anterior Pituitary >GI |
|
|
|
What occurs in MEN II?
|
>Endocrine neoplasia of the neural crest derived APUDs
|
|
|
|
How does vitamin D3 circulate in the bloodstream?
|
>Bound to vitamin D binding protein which has only a low affinity for the polar calcitriol
- this binding protein relationship is degraded after endocytosis in the renal tubular cells |
|
|
|
Where is calcitonin alternatively spliced? What is its alternative molecule?
|
>Spliced in the thyroid gland and in the brain
- where one transcript encodes calcitonin, the other encodes 'calcitonin gene-related peptide - CGRP acts as a neurotransmitter and a potent vasodilator |
|
|
|
What causes secretion of calcitonin?
|
>Plasma calcium concentrations of 2.1-3.0mmol Ca++
|
|
|
|
Name two mechanisms via which PTH increases the plasma calcium concentration
|
1. Bone resorption by osteoclasts
2. Activation of 1 alpha hydroxylase |
|
|
|
Via which second messenger pathway does PTH act?
|
cyclic AMP
|
|
|
|
What is the molecular basis for pseudohypoparathyroidism?
|
Receptor insenstivity
|
|
|
|
How does pseudohypoparathyroidism and idiopathic hypoparathyroidism differ?
|
>Clinical symptoms are the same (hypocalcaemia)
- The notable exception is in PTH levels which would be elevated in pseudohypoparathyroidism and absent / low in hypoparathyroidism |
|
|
|
What effect does PTH exert on the concentration of phosphate in the blood and urine?
|
>More phosphate in the urine, less in the blood (PTH increases the level of free / unbound Ca++ in the blood)
|
|
|
|
What is the major action of calcitriol?
|
>Acts by incerasing expression of an intestinal Ca++ transporter
- thereby increases intestinal uptake of Ca++ |
|
|
|
Upon which bone cells does PTH act on?
|
Osteoblasts
|
|
|
|
Upon which bone cells does calcitriol act on?
|
Osteoblasts
|
|
|
|
Upon which bone cells does calcitonin act on?
|
Osteoclasts
|
|
|
|
How many parathyroid glands are there usually?
|
4
|
|
|
|
Which proportion of parathyroids occur in an aberrant location?
|
10%
|
|
|
|
How many amino acids are encoded by the ORF for PTH? How many of these aas are required for the biological activity of PTH? At which end of the peptide chain?
|
>84 encoded, 34 needed, present at the amino terminal end of the chain
|
|
|
|
Which ions are important for PTH secretion from the chief cells?
|
>Ca++ most important (surface Ca++ receptor on chief cells)
>Mg++ also required for secretion |
|
|
|
How is calcitriol formed from vitamin D3?
|
>Maximises biological activity
- sequential hydroxylations must occur at C25 and at C1 (liver and kidney respectively) - enzyme responsible = 1 alpha hydroxylase |
|
|
|
Which enzyme inactivates vitamin D3?
|
24 hydroxylase
|
|
|
|
List 3 factors other than calcium which influence 1 alpha hydroxylase activity.
|
1. PTH (increases activity)
2. Calcitonin (decreases / inhbits) 3. Decrease in serum phosphate (increases activity) |
|
|
|
What are the symptoms of hypercalcaemia?
|
- Stones (renal or biliary)
- Bones (bone pain) - Groans (abdominal pain, nausea and vomiting) - Thrones (sit on throne - polyuria) - Psychiatric overtones (Depression 30-40%, anxiety, cognitive dysfunction, insomnia, coma) |
|
|
|
What causes are there are of hyperparathyroidism?
|
PRIMARY HYPERPARATHYROIDISM
- benign tumour of one or more parathyroid glands SECONDARY HYPERPARATHYROIDISM - compensation for long standing hypocalcemia - poor calcium intake / absorption - response to low calcitriol in chronic renal failure results in sub periosteal bone resorption - leading to cyst formation and bone pain |
|
|
|
What are possible causes of hypoparathyroidism?
|
IATROGENIC
- accidental removal of parathyroid glands during thyroid surgery IDIOPATHIC - no known cause, low circulating PTH, hypocalcemia and hyperphosphatemia PSEUDOHYPOPARATHYROIDISM - tissue resistance to PTH action - defect of bone and kidney PTH receptors - high circulating PTH, hypocalcaemia and hyperphosphatemia |
|
|
|
What does hypocalcaemia cause?
|
>Increased excitability of nerve tissue
- paraesthesia, tetany and sometimes epilepsy >Treatment by calcium supplements and calcitriol |
|
|
|
Why is hypercalcaemia a feature of malignant disease?
|
1. Malignant cells in bone secrete osteoclast activating factors, increasing bone resorption
2. Cancerous cells in bone are also associated with release of a PTH related peptide - a hypercalcaemic hormone with a structure similar to that of PTH |
|
|
|
What rare causes of hypercalcaemia exist?
|
>Excess calcitriol production
>Vitamin D overdose |
|
|
|
How would a medullary carcinoma of the thyroid affect calcitonin, PTH and Ca++ levels?
|
>Calcitonin would be elevated
>PTH and Ca++ would be normal - calcitonin, although is associated with reducing Ca++ levels, does not have a very strong effect over long term release of calcium |
|
|
|
What medical conditions result from Vit D deficiency?
|
Adult - osteomalacia
Child - rickets |
|
|
|
Why do the skeletal consequences of vitamin D deficiency differ with age?
|
>Depends on whether or not bones are still growing
|
|
|
|
In the face of adequate dietary vitamin D intake and exposure to UV light, why might the levels of calcitriol be insufficient to support intestinal calcium absorption?
|
>Renal or hepatic failure
- functional deficiency of vitamin D related enzymes, preventing sufficient calcitriol production |
|
|
|
Why might a respiratory alkalosis cause hypocalcaemia?
|
>Increased exhalation of CO2 favours dissociation of carbonic acid which decreases concentration of protons in the plasma
- hypocalcaemia ensues because of the increased association between Ca++ and albumen which has become unbound from H+ >Thus hypocalcaemia doesn't always reflect defects in the endocrine axis controlling calcium homeostasis |
|
|
|
In which foods is vitamin D found?
|
>Oily fish, herring, salmon etc
>Cod and halibut liver oils >Butter, cream and cheese >Fortified breakfast cereals |
|
|
|
In which locations does vitamin D act to produce its hypercalcaemic effect?
|
Stimulation of INTESTINAL calcium absorption
- rapid action by recruitment of calcium transporters to cell surface - slow action by induction of calbindin D (calcium binding protein) Stimulation by reabsorption of calcium in the DISTAL RENAL TUBULE Stimulation of OSTEOBLASTS to secrete osteoclast stimulating factors - hence mobilisation of bone mineral |
|
|
|
How does PTH exert its intracellular effects?
|
>Via cell surface receptors on Obs and on the antiluminal surface of the distal renal tubules
- linked to cAMP and adenylate cyclase - cAMP released from renal cells and appears in blood and urine RAPIDLY after PTH administration - phosphaturic response peaks at 1.5 hours afterwards |
|
|
|
What controls secretion of PTH? Via which pathway?
|
>Serum Ca++
- parathyroid chief cells have a cell surface Ca++ receptor - linked to PLC, IP3 and DAG - hypercalcaemia inhibits secretion - hypocalcaemia stimulates secretion - Mg++ is required for secretion |
|
|
|
Where is calcitriol synthesised?
|
>Only in the PROXIMAL RENAL TUBULE
|
|
|
|
How does Calcitriol exert its effect intracellularly?
|
>Bound to Gc globulin it is endocytosed
- liberated via hydrolysis to bind to the VDR, causing a conformational change >Activated VDR forms a heterodimer with occupied or unoccupied retinoid X receptor - phosphorylation of an occupied RXR-VDR heterodimer permits binding to HRE on DNA and initiates transcription NB. in the absence of ligand the heterodimers act as repressors, too high levels of vitamin A causes formation of RXR homodimers which also repress transcription |
|
|
|
What is the main storage form of vitamin D in the body?
|
Calcidiol
|
|
|
|
How much vitamin D is enough?
|
Between 10-50 years 5 ug
50-70 10ug >70 years 15ug - however there is evidence that even higher levels of intake are desirable |
|
|
|
What are the non calcium actions of calcitriol?
|
INSULIN SECRETION
- via calbindin induction in pancreatic B islet cells - hence a possible role in diabetes mellitus and metabolic syndrome INHIBITION OF CELL PROLIFERATION AND INDUCTION OF DIFFERENTIATION - via GPCRs linked to phospholipase C, hence roles in cancer prevention, immune system function, adipocyte differentiation POTENTIATION OF APOPTOSIS INDUCED BY VITAMIN A - hence again a role in cancer prevention |
|
|
|
A mutation in which enzyme may cause hypercalcaemia in individuals taking recommended levels of vitamin D?
|
>Calcidiol 24 hydroxylase (inactivator)
|
|
|
|
In which locations does PTH act to produce its hypercalcaemic effect?
|
1. Increases bone resorption by stimulating OSTEOBLASTS to produce osteoclast activating factors
2. Increases RENAL TUBULE absorption of calcium - Ca++ excretion may increase because of the the greater filtered load due to hypercalcaemia induced by PTH 3. Increases phosphate excretion, lowering serum phosphate and so raising serum calcium |
|
|
|
What is the rate determining step in the synthesis of all steroid hormones?
|
>The conversion of cholesterol to pregnenolone
- catalysed by the enzyme cytochrome P450 cholesterole side chain cleavage - occurs in the mitochondria |
|
|
|
Why does the mitochondrial location of conversion of cholesterol to pregnenolone impose a further rate limitation on steroidogenesis?
|
>Because of the need for two receptors required to transport the pregnenolone out of the mitochondria which have short half lives
- stAR and PBRs |
|
|
|
What are the three distinct zones of the adrenal cortex?
|
1. Zona glomerulosa
- Aldosterone (C21) 2. Zona fasciculata - Cortisol (C21) 3. Zona reticulata - DHEA (dehydroepiandrosterone) |
|
|
|
What is reponsible for the zone specific pattern of steroid biosynthesis?
|
>The differential expression of key steroidogenic enzymes in each adrenocortical zone
>Two major enzymes are required 1. Hydroxysteroid dehydrogenases 2. Cytochrome P450 enzymes |
|
|
|
What regulates the synthesis of glucocorticoids and adrenal androgens?
|
>The hypothalamo-pituitary-adrenal axis
- hypothalamus releases CRH which stimulates the - anterior pituitary to release ACTH which stimulates the production of - cortisol and DHEA CORTISOL EXERTS THE NEGATIVE FEEDBACK EFFECT TO CONTROL THIS AXIS |
|
|
|
What is the peptide precursor for ACTH?
|
Pro opiomelanocortin (POMC)
|
|
|
|
How is the solubility of cortisol in plasma increased?
|
>Through corticosteroid binding globulin
|
|
|
|
What largely controls the synthesis of aldosterone?
|
>Renin-Angiotensin Axis
|
|
|
|
What would cause aldosterone synthesis to increase?
|
>Increase in Plasma [K+], decrease in plasma volume, increase in ACTH, increase in angiotensinogen
|
|
|
|
What would cause aldosterone synthesis to increase?
|
increase in plasma angiotensin II, ACTH, or potassium levels, which are present in proportion to plasma sodium deficiencies. (The increased potassium level works to regulate aldosterone synthesis by depolarizing the cells in the zona glomerulosa, which opens the voltage-dependent calcium channels.) The level of angiotensin II is regulated by angiotensin I, which is in turn regulated by the hormone renin.
- potassium levels are the most sensitive stimulator of aldosterone Other factors include: - plasma acidosis - adrenoglomerulotropin |
|
|
|
In what manner does cortisol cause hyperglycaemia?
|
>Through increasing expression of:
- glucose-6-phosphatase - fructose-1,6-bisphosphatase - PEP carboxykinase in order to stimulate gluconeogenesis |
|
|
|
How does cortisol produce sufficient substrate for gluconeogenesis?
|
>Via lipolysis of TAG to release glycerol and then subsequent beta oxidation of fatty acids
NB. not possible for NEFAs to serve as a gluconeogenic substrate |
|
|
|
What are the non-metabolic actions of glucose?
|
- increased inotropy
- increased GFR, renal blood flow and free water clearance - wound healing inhibition (fibroblast activity) - increased surfactant production (used as a treatment in RDS) - immune / antiinflammatory effect - catabolic action on bone, increasing circulating Ca++ |
|
|
|
Why are the adrenal androgens of little consequence in a healthy male?
|
>Because the testes produce androstenedione and testosterone which support the development of secondary sexual characteristics
|
|
|
|
What is the importance of adrenal androgens in pre menopausal women? Clinically why may they be important in post-menopausal women?
|
>Required at adrenarche to stimulate the growth of hair and pubic hair
- in a post menopausal female, adrenal androgens can be metabolised to oestrogens which may support the growth of oestrogen dependent tumours |
|
|
|
How would a decrease in ACTH affect aldosterone secretion?
|
There would be no change, as aldosterone is coupled predominantly to renin, and not to ACTH, although ACTH does stimulate its production
|
|
|
|
What is the structure of the adrenal gland?
|
>Small pyramidal / triangular shaped glands (4-6g in an adult), superior to the kidneys
>Consist of - medulla, where catecholamine synthesis takes place - cortex, 90% of the gland where corticosteroid synthesis takes place |
|
|
|
What is the sructure of the adrenal medulla? When do chromaffin cells develop?
|
>Comprised of columnar, chromaffin cells (affinity to chromium stain)
- progenitors of chromaffin cells are phaeochromaffinoblasts, derived from neural crest cells - week 7 of development phaeochromaffinoblasts colonise centre of developing adrenal cortex to establish medulla - embryonic adrenal cortex at anterior of mesonephroos, originates from mesothelium, invading retroperitoneal mesenchyme at week 5 of development |
|
|
|
What innervation do chromaffin cells have?
|
>Each chromaffin cell is innervated by a cholinergic, pre-ganglionic sympathetic neurone
- derived from splanchnic nerves (exiting the CNS between T8 and T11) - some chromaffin cells form paraganglia - collections of chromaffin cells scattered in retroperitoneal and retropleural sites (the function of these are known, but can rarely give rise to phaeochromocytomas) |
|
|
|
What is the blood supply to the adrenal gland?
|
>Supplied via superior, middle and inferior adrenal arteries
>Give rise to medullary arterioles, cortical and capsular vessels - since cortical capillaries contact the medulla, this receives a dual blood supply and steroids from the adrenal cortex delivered directly to the medulla >Blood exits via medullary venules, thence into central veins - right adrenal vein connects to the inferior vena cava, central vein from left adrenal empties into the left renal vein |
|
|
|
What is synthesised in chromaffin cells?
|
>Adrenaline
>Noradrenaline |
|
|
|
How are the catecholamines produced in chromaffin cells?
|
>Synthesised in a 4:1 ratio (norad. to ad)
- by the action of PNMT = phenylethanolamine-N-methyl transferase - PNMT stimulated by sympathetic nervous system and glucocorticoids (absence of PMNT from neurons means they can only synthesise noradrenaline - a potent neurotransmitter) |
|
|
|
What is the chromaffin cell cytoplasm rich in?
|
>Secretory granules containing catecholamines, met and leu enkephalins (opiates) synthesised within the medulla
- release can be stimulated >Granules are released in response to Ach |
|
|
|
What impedes the synthesis of chromaffin granules?
|
>Inhibition of tyrosine hydroxylase (e.g. with alpha methyltyrosine)
>Suppressing release and / or action of Ach e.g. with hexamethonium |
|
|
|
Describe the synthetic pathway for catecholamine synthesis.
|
|
CATECHOLAMINE SYNTHESIS
|
|
|
Upon which receptors do catecholamines exert their influence?
|
Hydrophilic therefore act via cell surface receptors
- alpha adrenoceptors - beta adrenoceptors |
|
|
|
To which second messenger pathway are alpha adrenoceptors coupled?
|
>PI-PLC and mobilisation of intracellular Ca++
|
|
|
|
To which second messenger pathway are beta adrenoceptors coupled?
|
>AC, cAMP, PKA
|
|
|
|
How does the extracellular component of the BAR differ from that of the AAR?
|
>Beta adrenoceptors have very short extracellular N termini, inadequate to serve as a ligand binding domain
- instead 7 transmembrane helices form a hydrophilic binding pocket which accommodates the ligand |
|
|
|
What may potentiate the action of the AC-cAMP-PKA pathway?
|
>Because cellular responses are mediated through the AC-cAMP-PKA pathway, actions are potentiated and or mimicked by increasing the half life of cAMP
- e.g. with caffeine, which inhibits hydrolysis of cAMP by cyclic nucleotide phosphodiesterases |
|
|
|
How do GPCRs typically become desensitised?
|
>Most GPCRs desensitise on continuous exposure to stimulating ligand (failure of signalling pathway to respond to stimulus despite continued presence of receptors at cell surface)
DESENSITISATION may be - homologous - cell ceases to respond to desensitising stimulus, but the same signal transduction pathway remains responsive to alternative signalling stimuli - heterologous - cell loses ability to respond to a particular signalling ligand following exposure to an alternative signalling stimulus, acting through a different receptors type |
|
|
|
What is the pathway for homologous sensitisation of the beta adrenoceptor?
|
>Relies on interaction with Gs via regions in
- 3rd intracellular loop - C terminal tail of the receptor >Prolonged activation: - beta adrenoceptor stimulates beta adrenoceptor kinase - catalyses phosphorylation of specific Ser and Thr residues in the third intracellular loop and C terminal tail - conformational changes in beta adrenoceptor - allows receptor to interact with beta arrestin (peripheral membrane protein) - subsequent interactions between desensitised receptor and Gs is blocked NB. Similar mechanisms implicated in desensitisation of all GPCRs, betaARK is only one family of G protein receptor kinases |
|
|
|
What are the actions of catecholamines?
|
NORADRENALINE
- increases both systolic and diastolic blood pressure (increasing mean BP) - increases plasma NEFAs - little effect on bronchial tone / gut motility - dilates pupil and induces piloerection ADRENALINE - adrenaline increases systolic and reduces diastolic (no change in mean BP) - decreases gut motility and can act as a bronchodilator - increases glycogenolysis rapidly which results in hyperglycaemia and lactic acidaemia (due to a high rate of glycolysis) - dilates pupil |
|
|
|
How are catecholamines metabolised? By which enzyme?
|
>Concentrations of catecholamines are decreased by postganglionic receptor mediated endocytosis
- then metabolised by monoamine oxidases to 3,4 dihydroxymandelate which can be reused as a neurotransmitter NON NEURONAL CELLS - catecholamines are metabolised by MAO and by catechol-O-methyl-transferase - act in reversible sequence to generate inert products, including 3-methoxy-4-hydroxymandelate (VMA) - cleared in urine - uptake is more important than metabolism in terminating the actions of catecholamines |
|
|
|
What occurs in adrenal medulla failure?
|
>Very rare
>Causes hypotension and hypoglycaemia >Replacement of adrenal corticosteroids is sufficeint to control the blood pressure and plasma glucose concentration following removal / destruction of the adrenal glands |
|
|
|
What might cause overproduction of catecholamines? What would be the main symptoms?
|
>Normally reflects presence of phaeochromocytoma or neuroblastoma
- often associated with tumours of same embryonic origin (MEN II - neural crest cell-derived) |
|
|
|
What are P450C11B-1 and 2?
|
>2 is a gene duplication of 1
>Two enzymes involved in corticosteroid synthesis P450C11B1 = 11 beta hydroxylase - hydroxylates 11-deoxycortisol in ZF P450C11B2 = aldosterone synthase - ""new"" 18 hydroxylase and aldehyde synthase functions - retains 11 beta hydroxylase role in ZG |
|
|
|
How does the fetal adrenal cortex differ from the adult?
|
contains the FETAL ZONE
- synthesises 16-hydroxy-androstenediol - converted to oestriol in the placenta by P450 AROM - atrophies in the neonate |
|
|
|
What are the metabolic actions of cortisol?
|
1. Inhibition of insulin mediated glucose disposal via GLUT4 receptors
2. Sparing glucose metabolism where possible 3. Increasing hepatic gluconeogenesis 4. Increasing availability of gluconeogenic substrates - e.g. glycerol (increased lipolysis) - free amino acids (protein catabolism) |
|
|
|
Which tissues are entirely reliant on glycolysis?
|
>Brain
>Erythrocytes >Renal medulla - most tissues are capable of using non-carbohydrates to derive reduced NADH / FADH2 for ATP synthesis - NEFA - beta oxidation - acetyl CoA (TCA) - amino acids - 6 routes into the TCA |
|
|
|
Can NEFAs be used for gluconeogenesis?
|
NO!
|
|
|
|
How is POMC processed?
|
>Expression of pre-POMC driven usually by CRH
- this cleaves a signal peptide to generate POMC >POMC cleaved (with PHC/endopeptidase) - pro-ACTH and beta-LPH |
|
|
|
Which other hormones do PHC/endopeptidase work on?
|
ACTH
MSH beta endorphins |
|
|
|
What is the difference between CBG and SHBG?
|
>Both steroid binding globulins
>Cortisol binding globulin / transcortin, only binds - C21 steroids >SHBG (sex hormone binding globulin) - can bind C19 and C18 steroids |
|
|
|
What are the non-metabolic actions of cortisol?
|
>Negative feedback of CRH and ACTH
>Positive ionotropic action on the heart, abnormal ECG >Increase renal blood flow, GFR, water clearance >Inhibits fibroblast proliferation / collagen formation >Increased bruising, impaired wound healing >Actions on mood and behaviour >Increased synthesis of surfactant >Anti inflammatory / immunosuppressant >Increase in serum calcium - bone catabolism |
|
|
|
In which diseases of the adrenals is there altered hormone synthesis?
|
1. Cushing's disease
2. Cushing's syndrome 3. Addison's disease 4. Adrenal insufficiency 5. Congenital adrenal hyperplasia 6. Primary hyperaldosteronism |
|
|
|
In which conditions are there altered hormone actions (adrenally)?
|
>Pseudohypoaldosteronism
>Glucocorticoid resistance >Apparent mineralocorticoid excess |
|
|
|
What are possible pathogeneses for abnormal adrenal function?
|
1. Increased / decreased endocrine drive
2. Aberrant expression and or activities of steroidogenic enzymes 3. Destruction of one or more adrenal zones (e.g. autoimmunity) 4. Defective steroid receptors e.g. specificity / binding / TAF activity 5. Iatrogenic syndromes |
|
|
|
What are ten symptoms of increased corticosteroid action?
|
1. Hyperglycaemia
- increased gluconeogenesis - increased glycogenolysis - increased protein catabolism 2. Insulin resistance (frank NIDDM) 3. Redistribution of body fat - increased lipolysis in extremities - increased central lipogenesis - moon face and buffalo hump and abnormal adiposity 4. Increased bruising - weakened vessel walls 5. Immune suppression - antiinflammatory action (PLA2) - lymphatic / thymic involution - antagonises cytokine action 6. Gastric ulceration (decreased gastric pH) 7. Osteoporosis 8. Hyperandrogenism - acne, male pattern baldness and hirsutism - female pseudohermaphroditism - ?precocious puberty 9. Electrolyte and fluid imbalance - hypokalemia - anti natriuresis 10. Hypervolemic hypertension - headaches, visual disturbances, stroke |
|
|
|
What are seven symptoms of decreased corticosteroid action?
|
1. Hypoglycaemia
- tiredness and weakness 2. Vomiting 3. Hyper pigmentation - particularly skn creases and mouth 4. Hypoandrogenism - symptoms only evident in females - failed adrenarche - sparse axillary and pubic hair growth 5. Electrolyte and fluid imbalance - natriuresis / hyponatremia (? neuropathy) - anti kaliuresis 6. Polyuria - leading to polydipsia 7. Hypovolemic hypotension |
|
|
|
What are the effects of acute vs. chronic stress on the adrenals?
|
>Acute stress - causes catecholamine release
>Chronic stress - causes glucocorticoid release Both have actions of elevating plasma glucose - altering carb metabolism - increasing fat / protein mobilisation - decreasing insulin sensitivity |
|
|
|
What are possible causes of increased ACTH drive?
|
1. Chronic stress (increased CRH)
2. Glucocorticoid resistance (loss of negative feedback) 3. CRH independent secretion of ACTH (pituitary - corticotroph tumour) 4. Ectopic ACTH secretion (e.g. small lung cell tumour) 5. Defect in cortisol synthesis (CAH or Addison's disease) |
|
|
|
What is the difference between Cushing's disease and Cushing's syndrome?
|
>Cushing's disease - pituitary defect
>Cushing's syndrome - increased cortisol action not due to a pituitary tumour - chronic stress - ectopic ACTH secretion - functional ZF tumour - NOT glucocorticoid resistance |
|
|
|
How may Cushing's be diagnosed?
|
Tests may include:
- collection of urine over a 24 hour period to test for levels (morning rise in cortisol to be expected) - dexamethasone suppression test in which a synthetic cortisol is taken overnight or over the course of several days and blood or urine cortisol levels are measured at specific intervals - x-rays, scans and other tests to determine whether there is a tumour in the pituitary or adrenal glands or elsewhere |
|
|
|
How may Cushing's disease be treated?
|
>In many Cushing's cases, tumours that require surgery can be removed with minimally invasive techniques such as laparoscopic adrenalectomy
>Treatment can also include the gradual withdrawal of cortisone type drug and drug treatment to suppress adrenal gland function |
|
|
|
Describe the diurnal pattern of ACTH release.
|
>Highest in the early morning, between 6am and 8am
>Lowest in the evening, between 6-11pm |
|
|
|
What is ACTH testing used to measure?
|
1. Issues with the adrenal gland e.g. insufficiency / overproduction resulting in high / low levels of secretion
2. Low levels could be caused by an issue with pituitary gland 3. Overproduction of ACTH - hypersecretion due to a secreting tumour 4. Testing for the correct dosing of corticosteroids |
|
|
|
What is a dexamethasone suppression test? What is it used for?
|
>Carried out when Cushing's is suspected
- night before, at 11pm patient swallows a pill containing 1mg dexa - next morning a sample is taken - cortisol level is less than 5mg/dL or <138nmol/L |
|
|
|
What is a CRH test?
|
>Used to distinguish between Cushing's disease and an ectopic source of ACTH
- CRH is administered intravenously and the cortisol response monitored - normally there is a rise in both ACTH and cortisol - in Cushing's disease (pituitary dependent) patients the response is exaggerated, and in ectopic ACTH syndrome there will be no response (tumours aren't sensitive to CRH) |
|
|
|
What is inferior petrosal sinus sampling used for?
|
>This test can be used to accurately distinguish pituitary and ectopic sources of ACTH causing Cushing's syndrome
- the principle of the test is to sample the blood from the petrosal sinuses draining the pituitary gland, to compare the levels of ACTH with those found in the peripheral blood >A petrosal:peripheral ratio of >2, indicating excess ACTH from the pituitary is necessary to diagnose Cushing's with confidence - accuracy can be improved using CRH stimulation to exaggerate the difference |
|
|
|
What may cause hypocortisolaemia?
|
1. Impaired steroidogenesis
2. Decreased ACTH drive - decreased hypothalamic CRH drive - decreased pituitary ACTH output - ACTH resistance |
|
|
|
What causes Addison's disease?
|
>Most often occurs when the body's immune system kills off the part of the adrenal glands that makes cortisol and aldosterone
>May form when adrenals are harmed by: - infections such as TB, HIV and other bacterial or fungal infections - cancer that has spread to the adrenal glands, mostly seen in lung cancer - bleeding into the adrenal glands as a side effect of using blood thinners - some types of surgery or radiation treatments - high doses of certain medicines e.g. ketoconazole - injury to the adrenals >May occur at any age >May occur if steroids are used long term and then suddenly not used |
|
|
|
How can an ACTH drive defect be differentiated from Addison's?
|
>Addison's - all adrenal steroids are suppressed
>ACTH defect - only ACTH dependent steroids are decreased, therefore - aldosterone is unaltered in the ACTH defect - it is decreased in Addison's leading to polydipsia, polyuria, hyponatremia, increased Na+ intake |
|
|
|
Which medical condition is characterised by decreased synthesis of adrenal corticosteroids?
|
Addison's
|
|
|
|
What occurs in CAH?
|
>Loss of function mutations / decreased expression of adrenal steroidogenic enzymes
>Little or no synthesis of cortisol or corticosterone >Decreased negative feedback >Elevated ACTH (stimulating hyperplasia) Regarding Aldosterone there are two forms of CAH - Salt wasting (insufficient synthesis of Aldosterone) - Salt sparing (simple virilising CAH), sufficient mineralocorticoid to stimulate Na+ resorption >Malignant hypertension may result if there is overproduction of 11-deoxycorticosterone (doesn't feedback normally to suppress ACTH and CRH, thereby being stimulated to overproduce and cause hypertension) |
|
|
|
Which gene is usually mutated in patients with salt-wasting CAH?
|
CYP21 (encoding P450 C21)
|
|
|
|
Which gene is typically mutated in salt sparing CAH?
|
CYP11B1
|
|
|
|
Why does the adrenal become hyperplastic in CAH?
|
>Due to inefficient cortisol production which results in increased ACTH secretion inducing overgrowth
|
|
|
|
Why do 46XX patients with CAH typically present with female pseudohermaphroditism?
|
>Due to excess in-utero DHEA production, causing virilsation of the outer third of genitals
|
|
|
|
What is a typical cause of iatrogenic Cushing's?
|
>Administration of synthetic glucocorticoids in asthma / chronic inflammation
|
|
|
|
Why must patients be weaned off of corticosteroids?
|
>To avoid an Addisonian crisis, due to shrinkage of cortisol producing cells
|
|
|
|
Which corticosteroid is responsible for symptoms of hyperglycaemia, gluconeogenesis, immune suppression, osteoporosis, insulin resistance, increased bruising, hirsutism, increased stomach acid / gastric ulceration?
|
CORTISOL
|
|
|
|
Which corticosteroid is responsible for symptoms of hypokalaemia, anti natriuresis and hypervolemic hypertension?
|
MINERALOCORTICOIDS
|
|
|
|
Which corticosteroid is responsible for acne, hirsutism and cliteromegaly?
|
ADRENAL ANDROGENS
|
|
|
|
Why does glucocorticoid resistance result in hyperpigmentation?
|
ACTH is overproduced whih can be converted into alpha MSH which stimulates melanocytes to produce melanin
|
|
|
|
How are Cushing's syndrome patients treated?
|
Resection of ectopic ACTH site
Adrenalectomy and steroid replacement Anti glucocorticoids e.g. RU486 (anti progestogen) |
|
|
|
Why does glucocorticoid resistance (or a defect in cortisol synthesis) result in a lack of hepatic glycogen?
|
>Cortisol increases plasma [glucose] via increased gluconeogenesis (enzymes and substrates)
>Cortisol increases hepatic glycogen as a glucose reserve for future use >Cortisol increases expression of glycogen synthetase |
|
|
|
Why does increased production of DOC in salt sparing CAH result in malignant hypertension?
|
1. DOC is produced in excess
2. Lack of (aldosterone and) cortisol raise ACTH drive, thus increasing DOC production 3. Normal feedback mechanisms do not control DOC production |
|
|
|
What is Conn's syndrome?
|
>Primary and secondary hyperaldosteronism are conditions in which the adrenal gland releases too much of aldosterone
- in secondary hyperaldosteronism, the excess aldosterone is caused by something outside the adrenal gland that mimics the primary condition >Primary hyperaldosteronism is caused by a benign tumour of the adrenal gland - thought to be cause of elevated BP in some patients >Secondary aldosteronism is generally related to high BP, related to cirrhosis of liver, heart failure, or over activity of the RAAS system e.g. a renin producing tumour |
|
|
|
How is Conn's treated?
|
Adrenalectomy
- only unilateral >Bilateral adrenalectomy associated with Nelson's syndrome (pituitary adenoma) - causing excess ACTH release - originally the treatment for Cushing's disease - caused muscle weakness and skin hyperpigmentation |
|
|
|
What stimulates release of chromaffin granules?
|
>Direct release
- histamine, serotonin, Ach mimetics (e.g. carbachol and nicotine) >Indirectly - via vasomotor centre of the medulla oblongata |
|
|
|
What is the structure and function of vasopressin compared with oxytocin?
|
>Both 1.1kDa nonapeptides, vasopressin is a gene duplication of oxytocin (evolved from it)
>Vasopressin - antidiuresis>milk ejection >Oxytocin - milk ejection>antidiuresis |
|
|
|
Where is vasopressin mainly synthesised?
|
>In the magnocellular neurones
- packaged into neurosecretory granules (with neurophysin) - transported along axons (supra-optic hypothalamic tract) - released from neuronal terminals in posterior pituitary gland via fenestrated capiliaries - inferior hypophyseal circulation. |
|
|
|
Where else is vasopressin synthesised?
|
>Some found in parvocellular neurones
- co secreted with CRH - potentiates CRH stimulation of ACTH - some ACP secretion occurs even if supraoptic / hypothalamic tract is damaged |
|
|
|
What controls vasopressin secretion?
|
NB. Osmolality is more effective than plasma volume (via angiotensin) but these synergise, i.e. if osmolality increases in concert with a loss of plasma volume, there is a steeper rise in vasopressin secretion.
|
CONTROL OF VASOPRESSIN SECRETION
|
|
|
What are the two key actions of vasopressin?
|
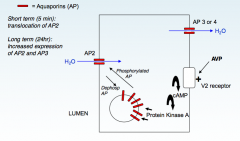
1. Pressor action - via V1 receptors (decreasing GFR)
2. Anti diuretic action - via V2 receptors - cAMP stimulates insertion of AQ2 water channels in renal collecting ducts (and longer term increased expression of AQ2 and AQ3 - Vasopressin is needed for Na+ -driven water resorption, but the t1/2 in plasma is approx 5 minutes, so longer term regulation of salt and water balance also involves aldosterone with a t1/2 of 20 minutes |
ADH EFFECTS
|
|
|
What are the actions of aldosterone on the renal distal tubule cells?
|
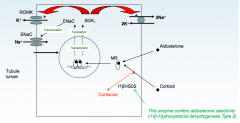
|
ALDOSTERONE EFFECTS
|
|
|
How is aldosterone secretion controlled?
|
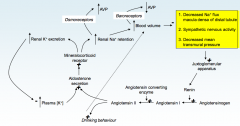
|
CONTROL OF ALDOSTERONE SECRETION
|
|
|
What is the structure of the adrenal cortex? What is produced in each layer of cortex?
|
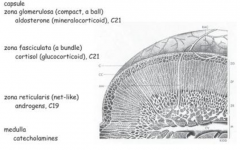
|
ADRENAL CORTEX
|
|
|
What are the possible causes of mineralocorticoid excess?
|
Primary - low renin, high aldosterone
1. Tumour of adrenal gland e.g. Conn's syndrome Primary - low renin, low aldosterone 1. Deoxycorticosterone producing tumour (usually adrenal) 2. Cushing's syndrome (but also diabetes mellitus) 3. Apparent mineralocorticoid excess Secondary - high renin, high aldosterone 1. Chronic haemorrhage/decrease in blood volume 2. Heart failure leading to decreased decreased glomerular filtration rate |
|
|
|
How may mineralocorticoid excess be treated?
|
1. Mineralocorticoid antagonists e.g. spironolactone
2. ACE inhibitors 3. K+ |
|
|
|
What are symptoms and possible causes of mineralocorticoid deficiency?
|
SYMPTOMS:
- hyponatremia / hyperkalemia, fatigue, weakness, hypotension 1. Addison's disease, autoimmune or infectious destruction of adrenal tissue 2. CAH (P450C21 or P450C11B2 deficiencies - enzymes on pathway to aldosterone) NB. It is NOT due to an acute decrease in ACTH - in fact it is often associated with pigmentation because of increased release of ACTH and melano |
|
|
|
What is diabetes insipidus?
|
PRESENTS WITH
- Polyuria (with decreased urine osmolality) - polydipsia (check not psychogenic) - increased plasma renin and aldosterone - hypovolemic hypotension CRANIAL - defect in AVP synthesis and or secretion NEPHROGENIC - defective V2 receptor protein expression |
|
|
|
Post injury to posterior pituitary / supraoptic hypothalamic tract, why isn't plasma vasopressin reduced to zero?
|
Vasopressin is co secreted with CRH from some parvocellular neurones
|
|
|
|
What treatment option is available for cranial / neurogenic DI?
|
>Vasopressin analogues e.g. desmopressin, in combination with a thiazide diuretic - for natriuresis)
|
|
|
|
How is nephrogenic DI treated?
|
>Extracellular fluid volume contraction via a low sodium diet and natriuretic thiazide diuretics. K+ may need replacing.
|
|
|
|
What occurs in SIADH?
|
>Hypersecretion of vasopressin associated with hypervolemia and potentially fatal hyponatremia (and production of concentrated urine)
- common disorder of fluid and electrolyte balance |
|
|
|
What are possible causes of SIADH?
|
>Could be due to
- intracranial trauma / infection - pneumonia - malignant disease e.g. SLCC - selected narcotics / analgesics e.g. nicotine, surgery - prolonged strenuous exercise e.g. marathon, hot weather training |
|
|
|
What are examples of vaopressin analogues?
|
>Porcine Lysine Vasopressin - low potency V2 receptor agonist
>Arginine vasopressin >Desmopressin (synthetic terminal cysteine deaminated and D rather than L arginine - high selective V2 receptor agonist >O ethyltyrosine substitute (synthetic) - vasopressin receptor antagonists |
|
|
|
What is the process which leads from an increase in plasma osmolality to secretion of vasopressin?
|
1. Increase in plasma osmolality
2. Increase in ECF osmolality 3. Osmotic cell shrinkage 4. Increased firing of osmoreceptors (hypothalamic and non hypothalamic) 5. Increased stimulation of magnocellular neurones 6. Increased secretion of vasopressin |
|
|
|
Which 3 major stimuli cause vasopressin secretion?
|
- Increased plasma osmolality
- Decreased plasma volume - Reduced O2 / increased CO2 |
|
|
|
Which are the two major inhibitors of vasopressin secretion?
|
>Oropharyngeal reflex
>Increased ANP (heart and CNS) |
|
|
|
What is the process which leads from an decrease in plasma volume to secretion of vasopressin?
|
1. Decrease in plasma volume
2. Decreased firing of baroceptors - systemic venous, left atrium, systemic arterial of carotid sinus and aortic arch 3. Decreased neurotransmission: afferents IX and X 4. Decreased inhibition (alpha adrenergic) of magnocellular neurones 5. Increased secretion of vasopressin |
|
|
|
What is more effective in controlling vasopressin secretion, osmolality or plasma volume?
|
Osmolality
|
|
|
|
What effect does a decrease in ACTH and an increase in angiotensinogen have on aldosterone secretion?
|
None
|
|
|
|
Why might plasma aldosterone be elevated in the presence of normal systemic arterial BP and normal plasma potassium?
|
Increased ACTH drive
|
|
|
|
What is Conn's syndrome?
|
>Primary hyperaldosteronism - RARE
>Increased secretion of aldosterone due to over-expression of steroidogenic enzymes and / or expansion of the zona glomerulosa >Increases kaliuresis / anti-natriuresis >Increases fluid resorption resulting in in hypervolemic hypertension >Secondarily causes a decrease in vasopressin concentration and decreased plasma renin activity |
|
|
|
What are possible causes of secondary hyperaldosteronism?
|
1. Decreased renal perfusion
2. Increased renin secretion 3. Increased angiotensinogen converted to angiotensin II 4. Increased aldosterone synthesis 5. Kaliuresis / Antinatriuresis 6. Increased fluid resorption - hypervolemic hypertension 7. Secondary fall in vasopressin concentration |
|
|
|
How can diet be relevant in hyperaldosteronism?
|
>Consumption of liquorice root which contains glycyrrhizin
- The most widely reported side-effects of glycyrrhizin use are hypertension and edema - These effects are related to the inhibition of cortisol metabolism within the kidney and the subsequent stimulation of the mineralocorticoid receptors - Thus, consumption of black licorice can mimic disorders of excess aldosterone |
|
|
|
How is Conn's diagnosed from secondary hyperaldosteronism?
|
>Plasma renin activity is increased in secondary hyperaldosteronism
(angiotensinogen isn't a rate limiting factor and is unaltered, plasma angiotensin II is too rapidly cleared to measure, urinary aldosterone is elevated in both, and urinary cortisol metabolites is irrelevant) |
|
|
|
What are other possible causes of hyperaldosteronism?
|
>Elevated ACTH
- Cushing's disease - Ectopic ACTH secretion >Hyperkalaemia |
|
|
|
What are the symptoms of pseudohypoaldosteronism?
|
>Natriuresis / Antikaliuresis
>Hyponatremia / Hyperkalemia >Increased plasma renin activity >Increased Aldosterone concentration Resistance to actions of aldosterone |
|
|
|
Name two aldosterone analogues.
|
9 alpha fludrocortisone
spironolactone (mineralocorticoid receptor antagonist) |
|
|
|
Why does increased production of DOC in salt sparing / simply virilising CAH result in malignant hypertension?
|
>DOC production insensitive to suppression of renin-angiotensin system and increase in plasma K+
|
|
|
|
What is the difference between type 1 and 2 corticosteroid receptors?
|
>Type 1 CR
- nominally the mineralocorticoid receptor - aldosterone and DOC are natural ligands - pharmacological ligands include DOCA and fludrocortisone >Type 2 CR - glucocorticoid receptor - cortisol / corticosterone = natural ligand - betamethasone, dexamethasone and prednisolone are pharmacological ligands |
|
|
|
What is the dilemma of the mineralocorticoid receptor?
|
>MR has no inherent specificity for mineralocorticoids
>The MR has a higher affinity than the GR - [Cortisol] > 100-1000 x [Aldosterone] - and cortisol displays a diurnal rhythm :. HOW DO MINERALOCORTICOID TISSUES RESPOND TO ALDOSTERONE RATHER THAN CORTISOL? |
|
|
|
How do mineralocorticoid receptors in the distal nephron, colon and parotid gland selectively respond to the RAAS?
|
>Because of the presence of 11 beta hydroxysteroid dehydrogenase type 2
- oxidises cortisol to 11-dehydrocortisol (or cortisone), greatly reduces activity at the mineralocorticoid receptor - the removal of two hydrogens converts the alcohol to a ketone, preventing the steroid from binding to either MR or GR (requires NAD+ coenzyme) |
|
|
|
What occurs in AME?
|
>The failure of the 11 beta hydroxysteroid dehydrogenase type 2 to protect the MR in the distal nephron and colon results in the syndrome of apparent mineralocorticoid excess (AME)
- clinical presentation of hyperaldosteronism but low plasma [aldosterone] with suppression of the RAAS >Enzyme fails to perform guardian role if mutated or inhibited |
|
|
|
Where do mutations typically occur in 11BHSD?
|
>In the coding sequence
>In promoter / response element (change level of expression - effects on regulation of gene expression) - recessive mutation :. AME is usually confined to consanguinous parentage - heterozygotes might have increased ability to conserve sodium under conditions of sodium deprivation giving a selective advantage which may have conserved mutations |
|
|
|
What may inhibit 11BHSD?
|
1. Environmental - liquorice, gossypol, glycyrrhetinic acid and bioflavinoids
2. Endogenous - bile pigments, sterols and steroids 3. Iatrogenic - carbenoxolone and frusemide |
|
|
|
If 11BHSD is fully active why might cortisol gain inappropriate access to MR?
|
- high cortisol e.g. Cushing's disease, glucocorticoid resistance
- exceeding capacity of enzyme |
|
|
|
What are the symptoms of AME?
|
>Anti natriuresis
>Alkalosis >Increased fluid resorption >Hypervolemic hypertension >Kaliuresis >Hypokalemia |
|
|
|
What is the role of placental 11BHSD?
|
>To protect the foetus from cortisol
>Failure causes: - increased passage of cortisol to foetus - stimulates premature differentiation of foetal tissues - prevents further growth of tissues - culminates in intrauterine growth retardation NB. Barker hypothesis, increased risk of serious adult disease (NB also obesity and type I enzyme) |
|
|
|
What is the function of 11BHSD type 1?
|
>It can reactivate glucocorticoids for actions at the MR
- overexpression of this enzyme particularly in fat can lead to symptoms similar to metabolic syndrome, and apparent glucocorticoid excess |
|
|
|
Where is 11BHSD type 1 typically found?
|
Ubiquitous - liver, lung, fat, gonad, pituitary is mostly type 1
|
|
|
|
What is a general phenomenon of target tissue steroid metabolism?
|
>Hydrophobic hormones are subject to metabolism to increase / decrease potency in target cells
|
|
|
|
Which endocrine glands are typically subject to an autoimmune attack?
|
>Thyroid
>Pancreas >Adrenal glands may also occur in the - gonads - pituitary NB. location of the target antigen that determines which endocrine glands will be attacked |
|
|
|
Where and how does tolerance for lymphocytes occur?
|
>Tolerance to self is normally established in the thymus for T cells and the bone marrow for B cells
- this occurs by clonal deletion (apoptosis) of any self reactive lymphocytes >Outside of the thymus, both T cells and B cells can be rendered ANERGIC (non responsive) as a back-up mechanism to ensure that they do not react to self antigens >ACTIVE SUPPRESSION by REGULATORY T CELLS of potentially autoreactive responses is yet another back-up mechanism NB. in autoimmunity this tolerance is lost and the immune system attacks itself |
|
|
|
What are examples of autoimmune thyroid diseases?
|
1. Hashimoto's disease
2. Atrophic thyroiditis (primary myxoedema) 3. Graves disease |
|
|
|
How commonly do autoimmune thyroid diseases occur?
|
>In 2-4% of the population, more commonly in females
|
|
|
|
What are likely causes of autoimmune thyroid diseases?
|
GENETIC:
- association with HLA DR3 and or HLA DR5 and with CTLA4 polymorphisms (CTLA4 is an off switch for T cells) - female:male ratio in Graves' disease is 7:1 and this predominance in females is partly due to the modulation of the autoimmune response by oestrogens (thyroid autoimmunity often subsides during pregnancy - high oestrogen levels) ENVIRONMENTAL: - a role for STRESS has been proposed in precipitating the disease, thus bereavement, divorce and job loss may proceed onset - this may involve neuroendocrine pathways influencing the immune system - SMOKING is weakly associated with Graves' disease and strongly associated with OPTHALMOPATHY - in areas of iodine deficiency, IODINE SUPPLEMENTATION can have the undesired effect of precipitating the development of Graves' disease |
|
|
|
On which genes are MHC I and II located?
|
>MHC I - A, B and C
>MHC II - DP, DQ and DR |
|
|
|
Where are MHC receptors found?
|
Only on PROFESSIONAL APCs
- B cells - dendritic cells - macrophages - thymic epithelium |
|
|
|
Why might polymorphisms in the HLA DR gene cause autoimmunity?
|
>HLA II DR gene products present antigenic peptides to CD4+ helper T cells
- polymorphic variants may present different autoantigenic peptides |
|
|
|
Why might polymorphisms in the CTLA4 gene cause autoimmunity?
|
>CTLA4 is a surface molecule on T cells which binds CD80 and CD86 (B7 and B7,1)
- CTLA4 switches off the T cell when bound (CD28 activates it) - polymorphisms may therefore influence T cell activation / inactivation |
|
|
|
What occurs in TID?
|
>Immunologically mediated destruction of the beta cells in the islets of Langerhans
- Islet cell antigens act as autoantigens in this disease |
|
|
|
What are the likely causes of TID autoimmunity?
|
GENETIC:
- susceptibility genes include HLA DQ8 - resistance genes include HLA DQ6 ENVIRONMENTAL: Infection - coxsackie virus - hepatitis C virus - echovirus Diet - wheat protein - soft drinks - eggs |
|
|
|
What is the autoimmune aetiology of Addison's disease?
|
>Autoimmunity against the adrenal gland, and autoantibodies against the enzyme 21 hydroxylase, as well as against cortical cell antigens are present
- there is atrophy of the adrenal gland leading to deficient production of adrenocortical hormones together with increased secretion of ACTH |
|
|
|
What is the prevalence of and F:M ratio in Addison's?
|
- 4:1
- 3-6 in 100000 |
|
|
|
What are the susceptiblity genes in Addison's?
|
>HLA DR3 and or DR4
|
|
|
|
What occurs in gonadal autoimmunity?
|
>Pathological autoimmunity can affect the gonads in either females (autoimmune oophoritis) or males (autoimmune orchitis)
- RARE occurrence and has not been very extensively investigated in patients from an immunological standpoint |
|
|
|
What is lymphocytic hypophysitis?
|
>Pituitary autoimmunity
- can be associated with pituitary hormone deficiency RARE - most often occurs in females, usually late in pregnancy or postpartum |
|
|
|
Which three major thyroid autoantigens are implicated in thyroid autoimmunity?
|
>Thyroid stimulating hormone (TSH) receptor
>Thyroid peroxidase >Thyroglobulin |
|
|
|
What are the autoimmune characteristics of Hashimoto's disease?
|
>Characterised by autoandtibodies to thyroid peroxidase and thyroglobulin (T4)
>Lymphocytic infiltration of the thyroid leads to destruction of thyroid tissue, causing hypothyroidism >Although the critical mechanisms are unclear it is thought that cytotoxic T cell attack is probably the main destructive mechanism, with complement-fixing autoantibodies also playing a role |
|
|
|
What are the autoantibodies in primary myxoedema?
|
>Blocking autoantibodies against the TSH receptor
>Prevent normal stimulation of the thyroid gland by TSH >Causes hypothyroidism |
|
|
|
What are the autoantibodies in Graves' disease?
|
>Stimulatory autoantibodies against the TSH receptor
>Cause hyperthyroidism >Hypersecretion of thyroid hormones, thyroid hypertrophy and hyperplasia of thyroid follicles |
|
|
|
What is the mechanism for autoimmunity in Hashimoto's disease?
|
1. Initiation of activation of naïve T helper cells by professional APCs which express both MHC class II and costimulatory molecules including CD80 and CD86
2. Thyroid cells aberrantly expressing MHC II can present thyroid antigens such as the TSH receptor, to activated Th cells (not to naive T cells however because thyroid cells lack the necessary co-stimulatory molecules to do this) 3. Presentation by thyroid epithelial cells therefore cannot initiate the disease, but may exacerbate the autoimmune response once it gets underway |
|
|
|
What are the typical steps which ensue following Th activation in autoimmune thyroid disease?
|
1. Activated Th1 cells secrete IL-2 and interferon y, aiding cell mediated immunity including activation of macrophages
2. Interferon y also causes thyroid cells to aberrantly express MHC II molecules (normally only present on professional APCs) 3. Activated Th2 cells secrete IL-6, IL-13 and other cytokines which help B cells make autoantibodies 4. Autoantibodies produced by thyroid infiltrating lymphocytes, with additional contributions from lymphocytes in cervical lymph notes and bone marrow |
|
|
|
What is the pathophysiology of Graves' opthalmopathy?
|
>Graves' disease often associated with opthalmopathy and pretibial myxoedema
>Enlarged extraocular eye muscles present in all Graves' patients if assessed by CAT scan >Infiltration of the extraocular muscles and orbital connective tissue by lymphocytes and macrophages >TSH receptor is expressed on orbital fibroblasts >They eye symptoms are probably mediated by T lymphocytes rather than by the anti TSH receptor autoantibodies >The inflammatory cells release cytokines including IL-1, TNF and interferon gamma, causing activation of fibroblasts ultimately leading to fibrosis |
|
|
|
What are the autoantibodies present in a type I diabetic?
|
>Glutamic acid decarboxylase
>Insulinoma-like antigen 2 >Insulin |
|
|
|
Which gene may confer susceptibility for TID?
|
HLA DQ8
|
|
|
|
Which gene may confer resistance to TID?
|
HLA DQ6
|
|